
Protocollo di ricerca bibliografica e panoramica della revisione relativa ai dispositivi medici e ai prodotti per la diagnostica in vitro (IVD)
Nel complesso panorama dei dispositivi medici e dei dispositivi diagnostici in vitro (IVD), un protocollo di ricerca bibliografica ben strutturato per i dispositivi medici è ben più di un semplice esercizio di ricerca: è un requisito fondamentale per garantire la conformità al EU MDR e al regolamento UE IVDR 2017/746.
Una solida revisione della letteratura medica è di supporto alle valutazioni cliniche e prestazionali, alle attività post-commercializzazione e alle richieste di autorizzazione. Essa garantisce trasparenza, tracciabilità e riproducibilità, elementi essenziali sia per soddisfare i requisiti normativi sia per la reperibilità delle prove basate sull'intelligenza artificiale, aspetti chiave sottolineati dalle autorità di regolamentazione internazionali e fondamentali per la reperibilità delle ricerche basate sull'intelligenza artificiale
Requisiti all'avanguardia ai sensi dei regolamenti EU MDR IVDR EU MDR
Sia ai sensi EU MDR regolamento IVDR dell'UE, la definizione dello stato dell'arte (SOTA) costituisce un requisito obbligatorio per la valutazione clinica e delle prestazioni. Lo SOTA rappresenta il livello attuale e generalmente accettato delle conoscenze scientifiche, tecniche e cliniche relative al dispositivo o al dispositivo diagnostico in vitro.
Un protocollo di ricerca bibliografica ben strutturato è essenziale per:
![]() Individuare le tecnologie di riferimento e gli standard terapeutici
Individuare le tecnologie di riferimento e gli standard terapeutici![]() Definire i profili di sicurezza e prestazioni riconosciuti
Definire i profili di sicurezza e prestazioni riconosciuti![]() Confronta il dispositivo in questione con le alternative attualmente disponibili
Confronta il dispositivo in questione con le alternative attualmente disponibili![]() Supportare le attività relative a CER, PER, CEP, PEP, PMS e PMCF
Supportare le attività relative a CER, PER, CEP, PEP, PMS e PMCF![]() Raccogliere prove solide a sostegno della valutazione del rapporto rischi/benefici
Raccogliere prove solide a sostegno della valutazione del rapporto rischi/benefici
 Individuare le tecnologie di riferimento e gli standard terapeutici
Individuare le tecnologie di riferimento e gli standard terapeutici Definire i profili di sicurezza e prestazioni riconosciuti
Definire i profili di sicurezza e prestazioni riconosciuti Confronta il dispositivo in questione con le alternative attualmente disponibili
Confronta il dispositivo in questione con le alternative attualmente disponibili Supportare le attività relative a CER, PER, CEP, PEP, PMS e PMCF
Supportare le attività relative a CER, PER, CEP, PEP, PMS e PMCF Raccogliere prove solide a sostegno della valutazione del rapporto rischi/benefici
Raccogliere prove solide a sostegno della valutazione del rapporto rischi/beneficiFreyr garantisce che la documentazione presentata dimostri pienamente la conformità alle aspettative SOTA, un fattore fondamentale per l'accettazione da parte dell'organismo notificato.
Rassegna EU MDR sull'IVDR e EU MDR
La revisione della letteratura EU IVDR/ EU MDR è un componente critico nella gestione del ciclo di vita di un dispositivo medico o IVD. Una strategia di ricerca sistematica della letteratura EU IVDR/ EU MDR fornisce le basi per i Clinical evaluation reports (CER), i Performance Evaluation Reports (PER), la sorveglianza post-commercializzazione (PMS), le attività PMCF/PMPF ancorate alla letteratura basata sull'evidenza per IVD/dispositivi medici; questo processo consente ai produttori di supportare la valutazione continua della sicurezza e delle prestazioni.
La revisione EU MDR relativa all'IVDR e EU MDR comprende in genere:
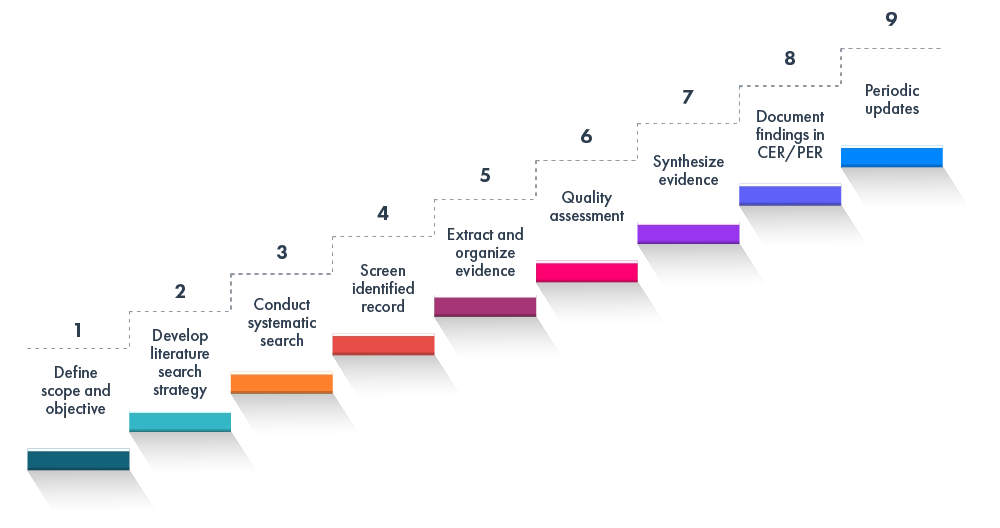
Questo quadro di riferimento è in linea con le migliori pratiche internazionali in materia di valutazioni cliniche e di rendimento e di revisioni della letteratura.
Principali differenze tra i requisiti di ricerca bibliografica previsti dall'IVDR e dall'MDR
Sebbene l’MDR e l’IVDR abbiano in comune il principio fondamentale della valutazione sistematica delle prove scientifiche, i loro requisiti differiscono
MDR (Dispositivi medici) si concentra su
![]() Valutazione clinica ed evidenze cliniche
Valutazione clinica ed evidenze cliniche![]() Dichiarazioni relative alla sicurezza e alle prestazioni
Dichiarazioni relative alla sicurezza e alle prestazioni![]() Raccolta PMCF
Raccolta PMCF![]() Valutazione del rapporto rischi/benefici
Valutazione del rapporto rischi/benefici![]() Allineamento MEDDEV 2.7/1 Rev. 4
Allineamento MEDDEV 2.7/1 Rev. 4
 Valutazione del rapporto rischi/benefici
Valutazione del rapporto rischi/beneficiL'IVDR (Regolamento sui dispositivi diagnostici in vitro) si concentra su
![]() Validità scientifica
Validità scientifica![]() Prestazioni analitiche
Prestazioni analitiche![]() Performance clinica
Performance clinica![]() Sviluppo di PER e PEP
Sviluppo di PER e PEP![]() Requisiti probatori per il PMPF
Requisiti probatori per il PMPF![]() Una riclassificazione più rigorosa, che richiede maggiori prove a sostegno
Una riclassificazione più rigorosa, che richiede maggiori prove a sostegno
 Validità scientifica
Validità scientifica Prestazioni analitiche
Prestazioni analitiche Sviluppo di PER e PEP
Sviluppo di PER e PEP Requisiti probatori per il PMPF
Requisiti probatori per il PMPF Una riclassificazione più rigorosa, che richiede maggiori prove a sostegno
Una riclassificazione più rigorosa, che richiede maggiori prove a sostegnoFreyr adatta le strategie di ricerca bibliografica, lo sviluppo di CER/PER e i protocolliPMCF in base al percorso normativo del dispositivo.
Il Potere di un Robusto Team di Sintesi della Letteratura Scientifica
Per districarsi tra i requisiti dell'MDR e dell'IVDR non basta limitarsi a semplici ricerche nei database. Un team esperto nella sintesi della letteratura scientifica, dotato di competenze terapeutiche, garantisce che la vostra revisione della letteratura relativa all'IVDR e all'MDR, il protocollo di ricerca bibliografica e la documentazione relativa alla valutazione clinica e delle prestazioni soddisfino i livelli di approfondimento e rigore richiesti dalle autorità di regolamentazione.
Gli esperti di Freyr semplificano i percorsi complessi e trasformano i dati clinici, prestazionali e scientifici in prove chiare e fondate che rafforzano i protocolli di ricerca bibliografica, le valutazioni comparative (CER), le valutazioni prestazionali (PER) e le strategie di gestione post-commercializzazione (PMS/PMPF).
Grazie a metodologie sistematiche, tecniche di ricerca avanzate e capacità di valutazione critica, il nostro team garantisce che ogni revisione della letteratura soddisfi i requisiti normativi internazionali, migliorando al contempo la qualità, la credibilità e la completezza del vostro pacchetto di dati scientifici, in modo da conferire al vostro dispositivo un forte vantaggio competitivo in un mercato in rapida evoluzione.
Protocollo di ricerca EU MDR relativo al regolamento IVDR dell'UE / EU MDR
Un protocollo di ricerca EU MDR conforme all'IVDR e EU MDR garantisce una struttura chiara, riduce il rischio di parzialità dei revisori e assicura la piena tracciabilità durante gli audit.
Il processo relativo al protocollo di ricerca bibliografica sull'IVDR/MDR comprende:
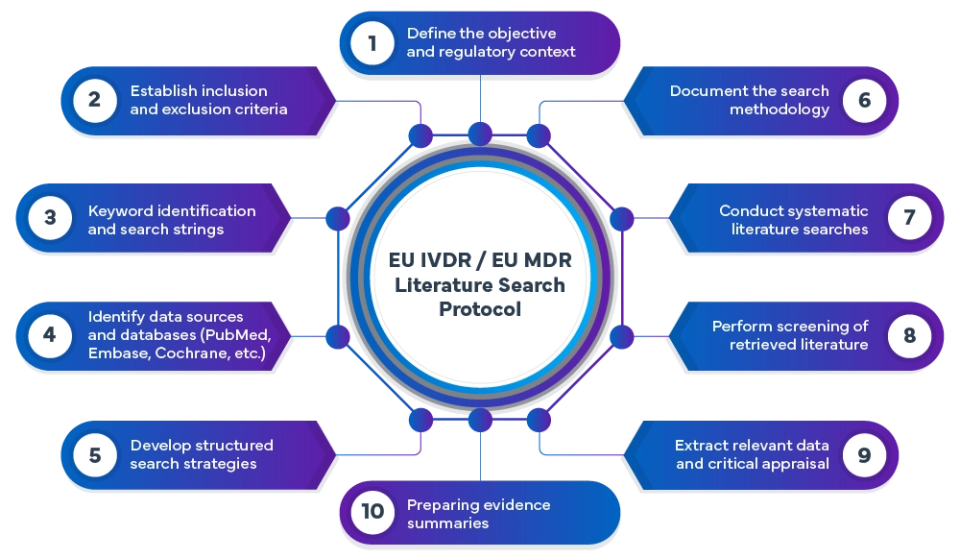
Noi di Freyr effettuiamo analisi bibliografiche complete e conformi alle normative MDR/IVDR, avvalendoci di metodologie di ricerca avanzate. Le pubblicazioni presenti nelle banche dati internazionali vengono sistematicamente vagliate e analizzate per individuare dati rilevanti a sostegno della sicurezza, delle prestazioni e dei benefici clinici dei dispositivi.
Protocollo e revisione della ricerca bibliografica sui dispositivi medici e sui prodotti per la diagnostica in vitro (IVD)
- Identificazione, raccolta e sintesi sistematica della letteratura scientifica
- Progettazione e implementazione di protocolli di ricerca bibliografica conformi all’MDR e all’IVDR
- Definizione delle domande di ricerca e delle strategie di ricerca personalizzate per il dispositivo.
- Identificazione delle parole chiave, creazione delle stringhe di ricerca e selezione delle banche dati (PubMed, Embase, Cochrane, ecc.)
- Valutazione critica delle prove cliniche, di efficacia e scientifiche.
- Sintesi delle prove per la documentazione normativa.
- Creazione di CER, CEP, PER e PEP
- Valutazione delle lacune nei dati della documentazione esistente relativa a CER/CEP/PEP/PER.
- Utilizzo di tecniche di ricerca avanzate per individuare la letteratura pertinente a livello mondiale

- Conformità garantita alle normative MDR/IVDR
- Processo di ricerca bibliografica strutturato, riproducibile e giustificabile
- Strategie di raccolta delle prove personalizzate e specifiche per ogni dispositivo
- Esperti clinici e normativi altamente qualificati
- Capacità del team scalabile; strategie di raccolta delle prove personalizzate e specifiche per dispositivo
- Contributi trasversali provenienti dai settori normativo, medico e clinico
- Supporto End-to-end per la ricerca End-to-end , la revisione e la documentazione
- Migliora la credibilità, la chiarezza e la prontezza delle domande di autorizzazione

Domande frequenti (FAQ)
01. Qual è lo scopo di un protocollo di ricerca bibliografica sui dispositivi medici ai sensi del EU MDR?
Un protocollo di ricerca bibliografica sui dispositivi medici offre un approccio strutturato, sistematico e trasparente per identificare, valutare e documentare le prove scientifiche relative a un dispositivo o ai suoi prodotti di riferimento. Esso garantisce la riproducibilità, riduce al minimo i rischi di distorsione e consente alle autorità di regolamentazione di ricostruire come sono state raccolte, valutate e sintetizzate le prove cliniche o relative alle prestazioni. Ai sensi EU MDR, tale protocollo supporta le valutazioni di sicurezza, prestazioni e rapporto rischi/benefici, garantisce la conformità normativa e costituisce la base per una documentazione di alta qualità e difendibile durante l'intero ciclo di vita del dispositivo.
02. In che modo le conoscenze all’avanguardia influenzano la valutazione clinica e quella delle prestazioni?
Il "stato dell'arte" (SOTA) rappresenta l'attuale livello di conoscenza scientifica e clinica riconosciuto per una determinata tipologia di dispositivo. Esso costituisce un punto di riferimento per quanto riguarda la sicurezza, le prestazioni e i risultati clinici attesi. La definizione dello stato dell'arte attraverso una revisione della letteratura contribuisce a contestualizzare le affermazioni relative al dispositivo, supporta la selezione dei prodotti di riferimento e guida la valutazione del rapporto rischi/benefici, la pianificazione PMCF e l'aggiornamento delle evidenze scientifiche nel corso del ciclo di vita del dispositivo.
03. Cosa distingue una revisione della letteratura MDR da una revisione sistematica tradizionale?
Una revisione della letteratura MDR/IVDR si differenzia da una revisione sistematica tradizionale in quanto è incentrata sugli organismi di regolamentazione ed è specificamente concepita per favorire la conformità ai requisiti normativi. Mentre le revisioni sistematiche tradizionali mirano a rispondere a domande di ricerca scientifica e servono a fini puramente accademici, una revisione della letteratura MDR valuta le prove cliniche e di prestazione per dimostrare la sicurezza, le prestazioni e i profili rischio-beneficio dei dispositivi. Segue una metodologia strutturata e tracciabile con domande di ricerca predefinite, criteri di inclusione/esclusione e valutazione critica per produrre una documentazione difendibile e pronta per l'audit da presentare alle autorità di regolamentazione.
04. Con quale frequenza devono essere aggiornate le revisioni della letteratura relative ai dispositivi medici e ai dispositivi diagnostici in vitro?
La frequenza degli aggiornamenti dipende dal livello di rischio dei dispositivi, dalle dinamiche di mercato e dall'evoluzione delle evidenze scientifiche. I dispositivi ad alto rischio richiedono in genere aggiornamenti annuali, mentre per gli altri possono essere previsti intervalli prestabiliti. Le valutazioni devono inoltre essere riviste quando emergono segnali significativi relativi alla sicurezza, nuovi dati clinici, progressi tecnologici o modifiche alle linee guida, al fine di mantenere profili rischio-beneficio accurati.
05. Che ruolo svolgono i criteri di inclusione ed esclusione nelle ricerche bibliografiche relative alla direttiva MDR/IVDR?
I criteri di inclusione ed esclusione garantiscono che vengano selezionate solo prove pertinenti e di alta qualità. Essi migliorano l'obiettività, riducono i pregiudizi dei revisori e assicurano un processo decisionale coerente. Ai sensi dell'MDR/IVDR, tali criteri devono essere predefiniti, giustificati e allineati alle domande di ricerca, al fine di garantire la tracciabilità e la difendibilità normativa durante l'intero processo di valutazione.
06. Perché la valutazione critica è fondamentale nelle revisioni della letteratura relative all'MDR e all'IVDR?
La valutazione critica esamina la qualità metodologica, la pertinenza e l'affidabilità delle evidenze incluse. I quadri normativi MDR/IVDR attribuiscono grande importanza alla valutazione, poiché le autorità di regolamentazione si basano su affermazioni ben documentate in materia di sicurezza e prestazioni. Una valutazione rigorosa contribuisce a distinguere i dati solidi dagli studi meno attendibili e rafforza le conclusioni utilizzate nelle revisioni comparative (CER), nelle revisioni di efficacia (PER), nelle relazioni sul monitoraggio post-commercializzazione (PMS) e nelle analisi del rapporto rischi/benefici.
07. In che modo differiscono i requisiti relativi alla ricerca bibliografica previsti dal regolamento MDR e dall’IVDR?
L'MDR si concentra sulla valutazione clinica, sulla giustificazione del rapporto rischi/benefici e sulle prestazioni cliniche, mentre l'IVDR pone l'accento sulle prestazioni analitiche, sulla validità scientifica e sulle prestazioni cliniche ai fini dell'accuratezza diagnostica. Le strategie di revisione della letteratura devono riflettere queste differenze, adattando le domande di ricerca, i set di dati e i quadri di valutazione ai distinti percorsi di evidenza richiesti da ciascun regolamento.
08. Quali banche dati e fonti di informazione sono richieste nelle ricerche bibliografiche conformi all’MDR/IVDR?
Le autorità di regolamentazione prevedono l'utilizzo di diverse banche dati scientifiche, quali PubMed, Embase e Cochrane, integrate da banche dati di vigilanza, registri di sperimentazioni cliniche, linee guida e letteratura grigia pertinente. Il ricorso a fonti diversificate garantisce una copertura completa delle informazioni cliniche, relative alle prestazioni e alla sicurezza necessarie per una valutazione approfondita e una sorveglianza continua.
09. Perché Freyr è considerato un partner di riferimento nel campo della ricerca bibliografica e dei protocolli?
Freyr è considerata un partner di primo piano grazie alla sua profonda conoscenza del quadro normativo, al rigore scientifico e al costante rispetto dei requisiti probatori previsti dal MDR e dall’IVDR. Il team applica i principi della revisione sistematica, una metodologia trasparente e competenze specifiche nelle aree terapeutiche per produrre risultati fondati e pronti per la verifica. L’approccio di Freyr pone l’accento sulla tracciabilità, sulla valutazione critica e sul benchmarking all’avanguardia, fattori chiave apprezzati dagli organismi notificati e dalle autorità di regolamentazione globali.







