
Perché la sorveglianza post-commercializzazione è importante nel ciclo di vita dei dispositivi medici
La sorveglianza post-commercializzazione (PMS) è un elemento fondamentale del ciclo di vita dei dispositivi medici, soprattutto alla luce dell’evoluzione normativa rappresentata EU MDR e dall’IVDR EU MDR e dai requisiti FDA . Con l’avanzare della tecnologia dei dispositivi, che spazia SaMD ai dispositivi indossabili e alle tecnologie connesse, il monitoraggio continuo nel mondo reale e l’individuazione proattiva dei rischi diventano essenziali. Le tendenze del settore, quali le evidenze del mondo reale (RWE), la vigilanza digitale e gli esiti riferiti dai pazienti, evidenziano la necessità di una valutazione continua della sicurezza, delle prestazioni e dei benefici clinici dei dispositivi. Nell'ambito delle moderne aspettative in materia di sorveglianza post-commercializzazione, produttori integrare sempre più spesso la rendicontazione delle tendenze, il rilevamento dei segnali e le valutazioni basate sul rischio.

Nonostante la sua importanza, molti produttori difficoltà nel soddisfare le aspettative relative al PMS. Fonti di dati frammentate, obblighi di segnalazione globali non uniformi, canali multilingue per la segnalazione dei reclami e un crescente controllo normativo creano complessità operativa. La preparazione di piani PMS, PMSR, PSUR e PMCF richiede competenze specialistiche, un'accurata interpretazione dei dati e un coordinamento interfunzionale. Lacune nell'analisi delle tendenze, nel rilevamento dei segnali, nella valutazione dei rischi per la salute (HHE) o nella segnalazione di vigilanza sui dispositivi medici possono portare a rilievi di audit, potenziali richiami o rischi per la continuità del prodotto.
Freyr affronta queste sfide con soluzioni end-to-end complete e end-to-end progettate per soddisfare i requisiti normativi globali. I nostri team combinano competenze normative, metodologie strutturate e supporto multilingue per ottimizzare la gestione dei reclami, la segnalazione di vigilanza e la documentazione PMS. Grazie a una comprovata esperienza SaMD di Classe I-III, IVD e SaMD , Freyr garantisce reportistica di alta qualità, conformità tempestiva e preparazione agli audit, rendendoci un partner affidabile per produttori di dispositivi medici produttori una gestione affidabile ed efficiente del ciclo di vita PMS.
Elementi chiave della sorveglianza post-commercializzazione dei dispositivi medici
Una sorveglianza post-commercializzazione (PMS) efficace combina una serie di attività interconnesse volte a monitorare la sicurezza dei dispositivi, le prestazioni cliniche, l'esperienza degli utenti e i rischi emergenti per tutta la durata di vita commerciale del prodotto. Questi elementi costituiscono il fondamento delle aspettative normative globali previste dal EU MDR, dal regolamento IVDR e dai requisiti FDA . Comprendere ciascun elemento è essenziale per garantire la conformità, migliorare le prestazioni dei dispositivi nel mondo reale e mitigare in modo proattivo i rischi per la sicurezza.
![]()
Gestione dei reclami e degli eventi avversi
Gestione strutturata della ricezione, dell'analisi e dell'andamento dei reclami al fine di individuare tempestivamente eventuali segnali di sicurezza, garantire un'escalation tempestiva e garantire la conformità a livello globale con i requisiti di segnalazione previsti FDA, EU MDR e dalle normative regionali in materia di vigilanza.![]()
Segnalazione di Vigilance e
Identificazione tempestiva, valutazione e segnalazione di eventi avversi e incidenti gravi alle autorità di regolamentazione globali, garantendo una supervisione continua della sicurezza e la conformità ai requisiti EU MDR previsti FDA e EU MDR .![]()
Ritiri, rettifiche e rimozioni
End-to-end delle azioni correttive sul campo, comprese la valutazione dei rischi, la valutazione dei rischi per la salute (HHE), le notifiche alle autorità di regolamentazione, la comunicazione e le verifiche di efficacia, al fine di garantire la sicurezza dei pazienti e tutelare la credibilità sul mercato.![]()
Piano PMS «
» (PMSP)
Un piano strutturato di sorveglianza post-commercializzazione che definisca le responsabilità, le fonti dei dati, i processi e i criteri di valutazione al fine di garantire un monitoraggio coerente e proattivo delle prestazioni del dispositivo durante tutto il suo ciclo di vita.![]()
PMSR, PSUR
e ePMCF
Relazioni previste dalla normativa che riassumono i dati post-commercializzazione, tra cui la relazione sulla sorveglianza post-commercializzazione (PMSR), gli aggiornamenti sul rapporto rischi/benefici e le attività di follow-up clinico volte a dimostrare la sicurezza e le prestazioni costanti del dispositivo.![]()
Analisi delle tendenze e dati provenienti dal mondo reale
Analisi dei modelli di reclamo e dei dati sulle prestazioni reali per individuare i rischi emergenti, sostenere le azioni preventive e migliorare l'affidabilità dei prodotti attraverso pratiche strutturate di rendicontazione delle tendenze.






Servizi di sorveglianza post-commercializzazione di Freyr
Prenota un incontro con i nostri esperti oggi stesso
Riflettori puntati sul successo: risultati concreti, storie vere
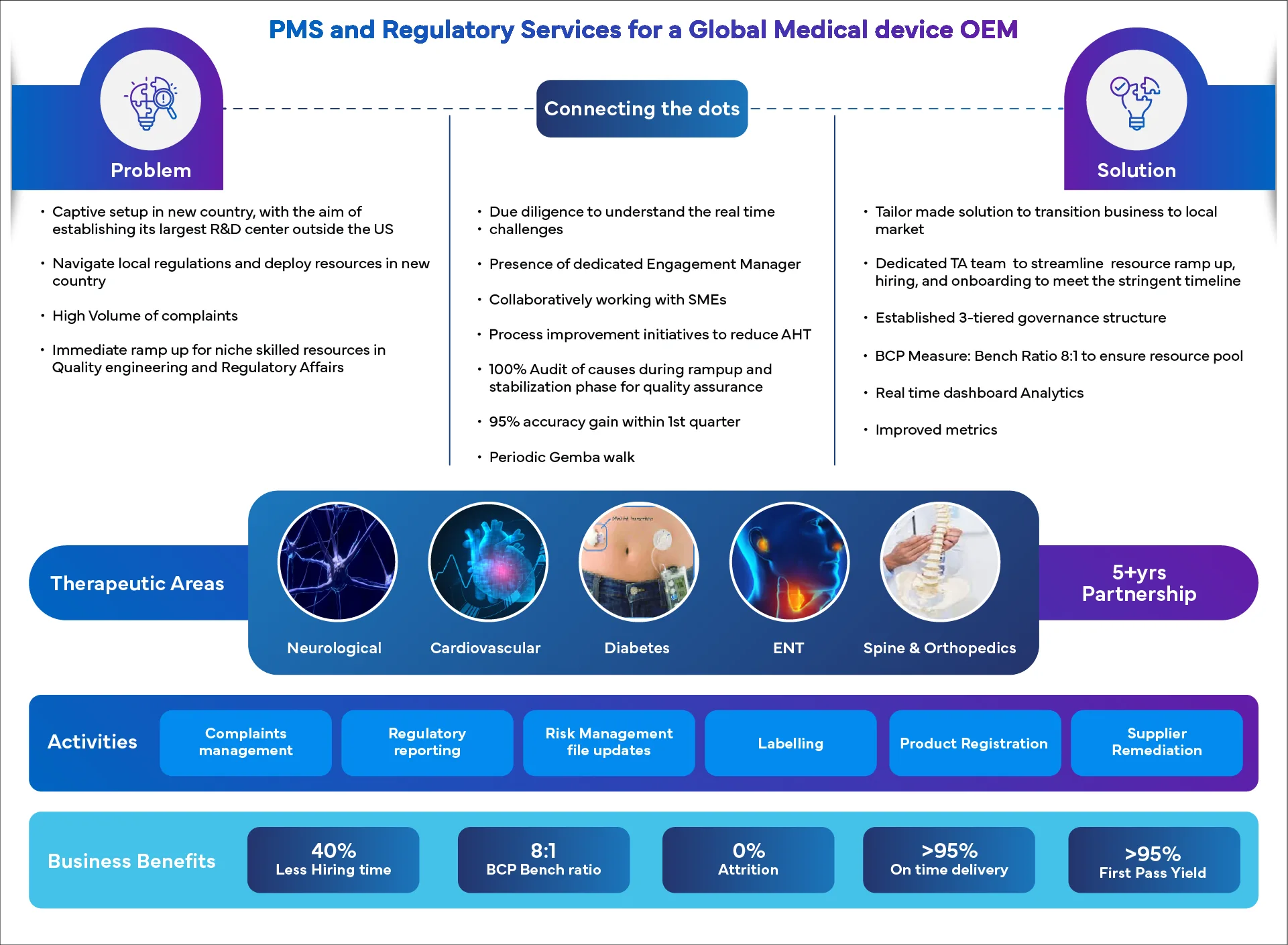
Perché collaborare con Freyr?
- La vasta esperienza maturata nell'ambito dei quadri normativi FDA, EU MDR, dell'IVDR e del PMS dell'APAC garantisce una conformità coerente e pronta per le ispezioni, nonché un solido allineamento normativo nei mercati globali.
- End-to-end , che spazia dalla gestione dei reclami alla segnalazione dei casi di vigilanza, passando per i rapporti PMSR, PSUR e PMCF l'intero ciclo di vita post-commercializzazione sia gestito con precisione ed efficienza.
- L'analisi basata sull'intelligenza artificiale, l'individuazione automatica delle tendenze e le informazioni ricavate dai dati reali aiutano a individuare tempestivamente i rischi emergenti, supportano la gestione delle azioni correttive e preventive (CAPA) e consentono di adottare decisioni proattive in materia di sicurezza.
- Gli esperti di lingua locale ottimizzano la selezione, la documentazione e l'invio dei reclami, garantendo un funzionamento fluido e accurato del sistema di gestione dei reclami (PMS) in tutte le regioni.
- Una comprovata esperienza nell'assistenza produttori gli audit FDA, EU MDR e degli organismi notificati, con una riduzione delle non conformità, migliori risultati in termini di qualità e una maggiore sicurezza in materia di conformità.
Domande Frequenti
01. Che cos'è la sorveglianza post-commercializzazione (PMS) nel settore dei dispositivi medici e perché è importante?
La sorveglianza post-commercializzazione è un processo continuo e sistematico volto a monitorare la sicurezza, le prestazioni e l'efficacia nella pratica clinica dei dispositivi medici dopo la loro immissione sul mercato. Si tratta di un processo essenziale in quanto consente produttori individuare i rischi emergenti, verificare nel tempo i benefici clinici, conformarsi alle normative globali in continua evoluzione e prendere decisioni basate su dati concreti per migliorare la qualità dei prodotti e la sicurezza dei pazienti.
02. Quali sono i requisiti fondamentali relativi ai sistemi di gestione della sicurezza (PMS) previsti dal regolamento EU MDR dal regolamento IVDR EU MDR ?
EU MDR il regolamento IVDR EU MDR impongono produttori predisporre un piano di sorveglianza post-commercializzazione (PMS), redigere periodicamente rapporti di sorveglianza post-commercializzazione (PSUR), svolgere PMCF quando necessario e istituire sistemi per la segnalazione dei casi di vigilanza e l'analisi delle tendenze. I regolamenti pongono l'accento sulla raccolta proattiva di dati, sull'integrazione dei dati clinici e sulla valutazione continua del rapporto rischi/benefici per tutto il ciclo di vita del dispositivo.
03. In che modo il PMS si integra con la gestione dei rischi e la norma ISO 14971?
Il sistema PMS integra direttamente le informazioni raccolte sul campo nel quadro di riferimento per la gestione dei rischi ISO 14971, consentendo una valutazione continua dei pericoli, delle modalità di guasto e dell'efficacia delle misure di mitigazione. I dati relativi a reclami, eventi avversi, rapporti sulle tendenze e follow-up clinico supportano inoltre la gestione delle azioni correttive e preventive (CAPA) e il miglioramento continuo.
04. Che ruolo svolgono i dati provenienti dal mondo reale (RWE) nella PMS?
I dati provenienti dall'esperienza clinica (RWE) rafforzano il sistema di monitoraggio post-commercializzazione (PMS) fornendo informazioni sull'uso effettivo dei dispositivi, tra cui il feedback dei pazienti, i dati di assistenza, i registri e le fonti di salute digitale. L'RWE contribuisce a identificare le tendenze, a convalidare le prestazioni a lungo termine, a supportare gli aggiornamenti sul rapporto rischi/benefici e a orientare le decisioni cliniche o normative. Le autorità di regolamentazione si aspettano sempre più spesso produttori integrino l'RWE nel PMS e nella valutazione post-commercializzazione continua.
05. Quando è necessario uno PMCF per un dispositivo medico?
Uno PMCF è richiesto quando le prove cliniche disponibili non sono sufficienti a confermare la sicurezza o le prestazioni a lungo termine, oppure quando la tecnologia, i materiali o la destinazione d'uso di un dispositivo suggeriscono potenziali rischi a lungo termine. PMCF inoltre avviato in presenza di nuove tendenze, rischi emergenti, incertezze cliniche, nuovi risultati derivanti dalla valutazione dei rischi per la salute (HHE) o quando le autorità di regolamentazione richiedono la raccolta continua di dati per dispositivi ad alto rischio o innovativi.
06. Quali sono i fattori che determinano il richiamo di un dispositivo medico o l'adozione di un'azione correttiva sul campo (FCA)?
Un richiamo o una FCA viene attivato quando un dispositivo presenta un rischio potenziale o accertato per la sicurezza, non soddisfa i requisiti normativi o manifesta problemi di prestazione che potrebbero compromettere gli esiti clinici. Tra i fattori scatenanti più comuni figurano tendenze relative a difetti, incidenti gravi, errori di etichettatura, difetti di fabbricazione, vulnerabilità nella sicurezza informatica o nuove prove che indicano un cambiamento nel profilo rischi-benefici del dispositivo.
07. Perché Freyr è considerata un partner di riferimento per i servizi di sorveglianza post-commercializzazione?
Freyr è ampiamente riconosciuta per la sua profonda competenza in materia normativa, la copertura dei mercati globali e l'approccio strutturato alla sorveglianza post-commercializzazione (PMS) in conformità con i requisiti EU MDR, FDA e dell'area APAC. Le organizzazioni apprezzano Freyr per la combinazione di conoscenze settoriali, metodologie basate sui dati, competenze multilingue e comprovata esperienza nel supporto a diversi tipi di dispositivi, che la rendono un partner affidabile per operazioni di sorveglianza post-commercializzazione affidabili e conformi.







