
Panoramica sulla registrazione dei dispositivi medici in Israele
L'industria dei dispositivi medici di Israele sta vivendo una crescita e un'innovazione sostenute, rendendola un centro per le tecnologie sanitarie all'avanguardia. La registrazione dei dispositivi medici è fondamentale per le aziende che entrano in questo mercato dinamico. Questa panoramica esplora gli aspetti chiave del processo di registrazione in Israele, offrendo approfondimenti sul quadro normativo e sui requisiti per portare dispositivi medici innovativi all'avanguardia del settore sanitario israeliano.
Autorità Regolatoria: La Divisione Dispositivi Medici del Ministero della Salute israeliano (AMAR).
Normativa: Legge sui dispositivi/apparecchiature mediche del 2012
Percorso Normativo: Registrazione del Prodotto
Rappresentante Autorizzato Locale in Israele: Titolare della Registrazione Israeliana (IRH)
Requisito SGQ: ISO 13485
Valutazione dei Dati Tecnici: Il Dipartimento Dispositivi Medici presso il Ministero della Salute
Validità della Licenza: Cinque anni
Formato di Presentazione: Cartaceo ed Elettronico
Traduzione: Documenti Tradotti in Ebraico
Classificazione del Dispositivo
La Legge e i Regolamenti sulle Attrezzature Mediche per la Registrazione delle Attrezzature Mediche in Israele non specificano un sistema di classificazione del rischio. Israele allinea invece la sua classificazione dei dispositivi medici agli standard internazionali, in particolare quelli delineati dai paesi del Global Harmonization Task Force (GHTF). In alternativa, la classificazione del rischio di un dispositivo in un paese riconosciuto viene adottata per la registrazione in Israele. Questo processo di classificazione considera tipicamente l'uso previsto, il livello di rischio e altri fattori che possono influenzare la sicurezza e l'efficacia dei dispositivi medici.
Classi dei Dispositivi Medici
| Classe | Rischio |
|---|---|
| Classe I | Basso |
| Classe II | Basso-Medio |
| Classe III | Elevato |
Modifiche proposte ai percorsi di registrazione.
Le modifiche proposte si applicano ai dispositivi di Classe I e Classe II, mentre il sistema di registrazione per i dispositivi di Classe III rimane invariato.
- I dispositivi di Classe I possono essere registrati immediatamente tramite autocertificazione.
- Per i dispositivi di Classe II, sebbene siano necessarie dichiarazioni e documenti tecnici, AMAR può accelerare il processo a quattordici giorni per quelli considerati a rischio medio-basso. Ciò si applica se il produttore detiene due autorizzazioni da paesi riconosciuti e fornisce sei mesi di dati di mercato. In alternativa, per i dispositivi di Classe II con solo l'autorizzazione US FDA 510(k) e sei mesi di dati di mercato US, il tempo di elaborazione AMAR è accelerato a sessanta giorni.
Rappresentante Autorizzato Locale in Israele
Le aziende di dispositivi medici con sede al di fuori di Israele devono nominare un Titolare della Registrazione Israeliano (IRH) per facilitare la registrazione dei loro prodotti destinati alla vendita nel paese. L'IRH funge da rappresentante locale del fabbricante ed è incaricato di mantenere i contatti con il Ministero della Salute per assicurare il rispetto delle normative locali. Inoltre, un IRH è responsabile di stabilire e mantenere una presenza commerciale in Israele, oltre a ottenere e conservare una licenza commerciale valida.
Registrazione dei Dispositivi Medici
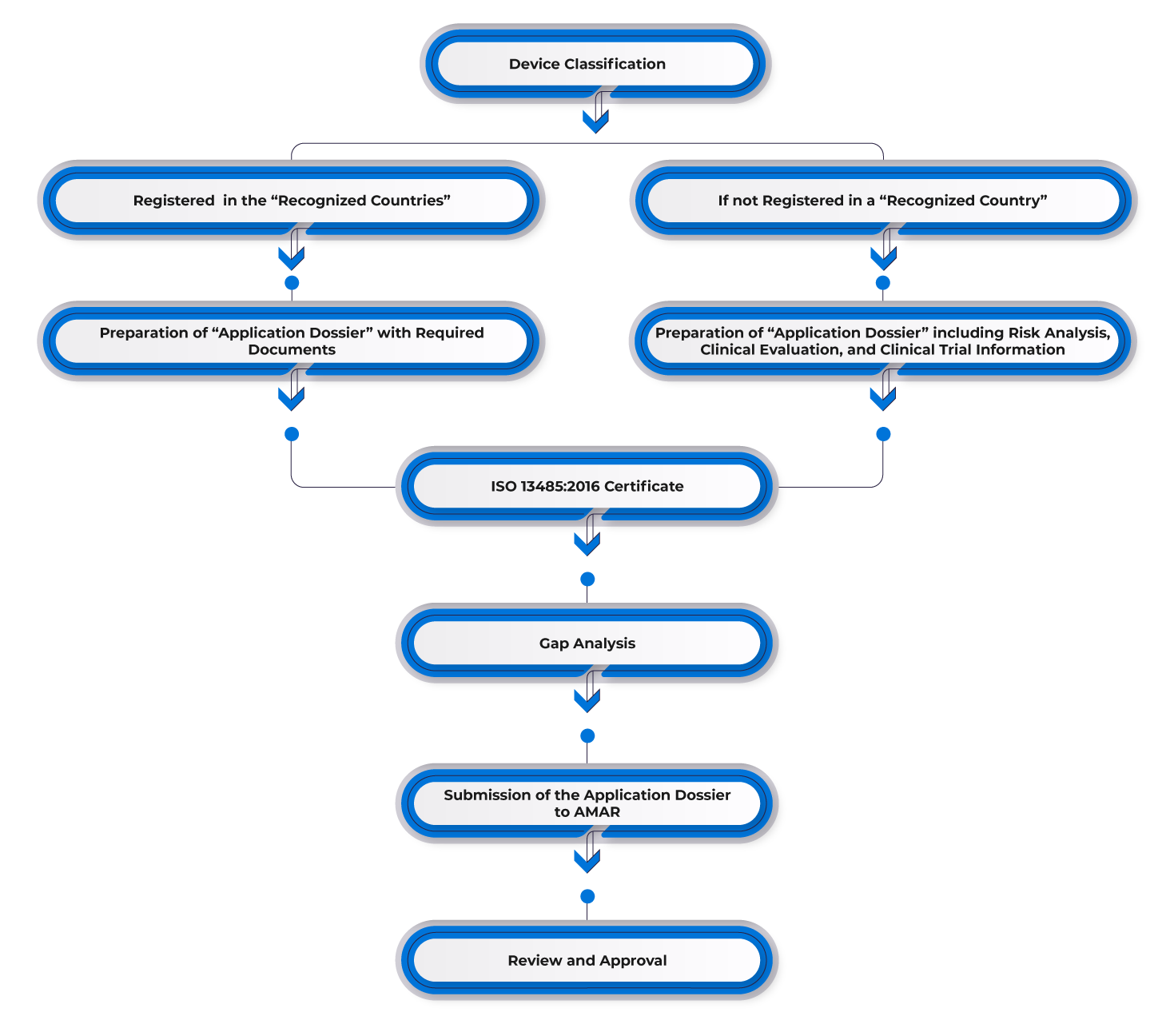
Per registrare un dispositivo medico in Israele, i produttori devono ottenere l'approvazione preventiva in uno dei mercati di riferimento come gli STATI UNITI D'AMERICA, l'Europa, l'Australia, il Canada o altri mercati importanti. I produttori con approvazioni esistenti in uno dei paesi di riferimento possono utilizzare questa approvazione per il mercato israeliano e nominare un rappresentante locale. Successivamente, devono presentare la documentazione richiesta, inclusi:
- FDA 510(k)/Lettera di approvazione pre-commercializzazione/CE.
- Certificato per il Governo Straniero (CFG)/Certificato di Libera Vendita (CFS).
- Certificazione ISO 13485 o un'altra certificazione riconosciuta di Buone Pratiche di Fabbricazione (GMP).
- Convalida e certificazione dall'Istituto Israeliano per gli Standard (se necessario).
Flusso del processo
Gestione del ciclo di vita dei dispositivi post-approvazione
Freyr offre un supporto completo ai produttori esteri nella gestione dell'intero ciclo di vita dei dispositivi medici in Israele, comprese le attività post-approvazione:
- Gestione delle modifiche post-approvazione, che riguardano le modifiche alle approvazioni esistenti dei dispositivi medici, come l'aggiunta di nuove varianti, accessori e indicazioni d'uso.
- Mantenimento della certificazione ISO 13485:2016 e CE.
- Rinnovo delle licenze.
- Agire come intermediario tra l'Organismo Notificato (NB) e il fabbricante.
Orientarsi nelle complessità degli organismi di autorizzazione e conformarsi a più insiemi di normative per le approvazioni dei dispositivi può essere impegnativo. Ottenere approvazioni da vari paesi GHTF e aderire alle normative statali richiede una conoscenza normativa approfondita. Per gli operatori che affrontano queste complessità senza un partner normativo consolidato, Freyr offre servizi normativi End-to-End, semplificando il processo di approvazione per i dispositivi medici in Israele.
Competenza nella registrazione dei dispositivi medici in Egitto
- Classificazione dei dispositivi medici in Israele.
- Titolare della Registrazione in Israele (IRH).
- Registrazione dei dispositivi in Israele.
- Consulenza sulla gestione del rischio ISO 14971:2019.
- Conformità ISO 13485:2016.
- Revisione, Compilazione e Presentazione del Dossier di Progettazione.
- Registrazione dei Dispositivi Medici tramite Sistema di Registrazione Online.
- Rapporto sulla Strategia Normativa dei Dispositivi Medici.
- Supporto ai test - Biocompatibilità, sicurezza elettrica, meccanica e prestazioni.
- Supporto alla conformità dell'etichettatura.
- Supporto GMP.
- Supporto alla Post-market Surveillance (PMS).
