
Panoramica sull'approvazione pre-commercializzazione dei dispositivi medici USFDA
Il processo di approvazione pre-commercializzazione (PMA) della USFDA è uno dei percorsi di registrazione dei dispositivi forniti dalla US FDA, progettato principalmente per i dispositivi medici di Classe III della FDA. Il processo di approvazione PMA della FDA per i dispositivi di Classe III comporta meticolose valutazioni scientifiche e normative per valutare la sicurezza e l'efficacia del dispositivo medico, garantendo che i più alti standard siano soddisfatti prima dell'autorizzazione all'immissione sul mercato.
Prenota un incontro con i nostri esperti in approvazione pre-commercializzazione.
Chi dovrebbe presentare una Domanda di approvazione pre-commercializzazione (PMA) per dispositivi medici USFDA?
I produttori di dispositivi devono presentare una domanda PMA se il dispositivo:
- È innovativo.
- Appartiene a una classe ad alto rischio.
- Non è presente nel database di classificazione dei prodotti.
- Non è sostanzialmente equivalente (NSE) a dispositivi di Classe I, II o III.
Ottieni consulenza esperta sulla tua domanda di approvazione pre-commercializzazione
Qual è la Differenza tra le domande 510(k), PMA e De-Novo?
Approvazione Pre-commercializzazione
- Dispositivo appartenente alla Classe III che supporta la vita umana o che presenta un rischio potenziale e irragionevole di malattia o lesioni.
- Il processo di approvazione PMA della FDA richiede studi clinici.
- Richiede un'ispezione in loco prima del rilascio dell'approvazione PMA.
- 180 giorni solari
Classificazione De Novo
- Dispositivi innovativi di Classe I e II che non hanno un dispositivo di riferimento valido.
- Richiede dati di studi clinici.
- Nessun audit in loco prima dell'approvazione De-Novo.
- 150 giorni solari.
Registrazione 510(k)
- Dispositivi FDA di Classe III che presentano un'equivalenza sostanziale con il dispositivo di riferimento.
- Non richiede test sull'uomo.
- Nessun audit in loco prima dell'autorizzazione 510(k).
- 90 giorni di calendario.
Quali sono i diversi metodi di richiesta di approvazione pre-immissione in commercio della FDA?
I produttori possono optare per uno dei seguenti quattro metodi di richiesta PMA che sarebbero più adatti per il loro dispositivo:
- PMA Tradizionale
- PMA Modulare
- Protocollo di Sviluppo del Prodotto
- Esenzione per dispositivi umanitari
Quali sono i requisiti di dati per l'approvazione pre-commercializzazione dei dispositivi medici?
Secondo il 21 CFR parte 814, i richiedenti devono presentare un modulo di domanda CDRH debitamente compilato, gli impegni richiesti e un fascicolo tecnico PMA ben redatto alla US FDA. Il fascicolo tecnico deve includere i dati non clinici e clinici.
Dati non clinici – Consiste in dati su microbiologia, tossicologia, immunologia, biocompatibilità, stress, usura, durata di conservazione e altri test di laboratorio o su animali.
Dati Clinici – Consistono in dati sui protocolli di studio, dati di sicurezza ed efficacia, reazioni avverse e complicazioni, guasti e sostituzioni dei dispositivi, informazioni sui pazienti, reclami dei pazienti, tabulazioni dei dati di tutti i singoli soggetti, risultati delle analisi statistiche e qualsiasi altra informazione derivante dalle indagini cliniche.
Cos'è il processo di domanda PMA?
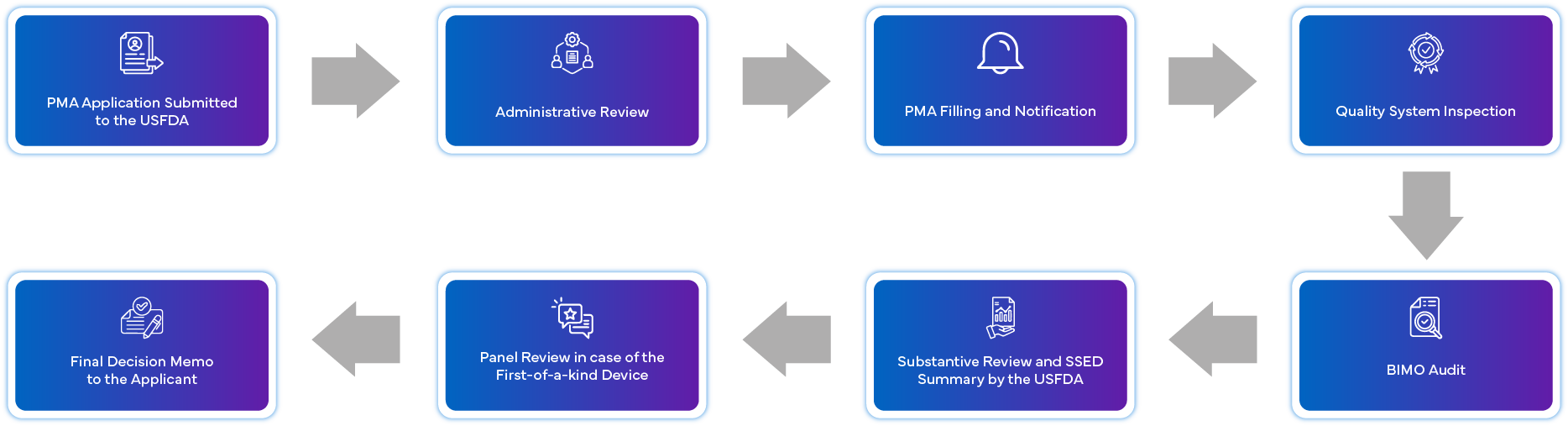
Quali sono i requisiti di conformità post-approvazione per PMA?
I dispositivi approvati tramite il percorso PMA devono essere conformi ai requisiti post-commercializzazione stabiliti dalla USFDA. Il dispositivo deve essere conforme a quanto segue:
- Requisiti post-approvazione imposti nell'ordine di approvazione PMA della FDA.
- Gestione delle modifiche post-approvazione tramite la presentazione tempestiva dei supplementi PMA pertinenti
- Sottomissione per i rapporti post-approvazione (annuali)
- Regolamenti 21 CFR 803 per la segnalazione dei dispositivi medici (MDR)
- Studi di Sorveglianza Post-commercializzazione come richiesto dalla USFDA negli ordini di approvazione PMA.
Quali sono le tariffe della USFDA per la revisione della domanda PMA?
Le tariffe utente MDUFA per PMA originali e supplementi sono le seguenti:
| Tipo di domanda | Tariffe per l'anno fiscale 2023 (dal 1° ottobre 2022 al 30° settembre 2023) | |
|---|---|---|
| Tariffa standard | Tassa per piccole imprese | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Supplemento Panel-Track | $353,238 | $88,309 |
| Supplemento di 180 giorni | $66,232 | $16,558 |
| Tassa annuale per la rendicontazione periodica su un dispositivo di Classe III (PMA, PDP e PMR) | $15,454 | $3,864 |
| Preavviso di 30 giorni | $7,065 | $3,532 |
| Integratore in tempo reale | $30,908 | $7,727 |
Con esperienza nella gestione delle sottomissioni PMA, Freyr può assistere nell'identificazione e nella compilazione delle informazioni e nella preparazione e revisione della domanda.
Competenza e vantaggi nell'approvazione pre-commercializzazione dei dispositivi medici USFDA
- Due Diligence Regolatoria
- Conformità all'Ispezione del Sistema Qualità
- Conformità agli Audit BIMO
- Compilazione del Fascicolo Tecnico PMA
- Pubblicazione e creazione di eCopy
- Convalida e presentazione dell'eCopy
- Gestisce la risposta RTA e le carenze
- Servizi di collegamento fino all'approvazione pre-commercializzazione della FDA.
- Consulenza sulle carenze
- Registrazione del dispositivo e registrazione dello stabilimento
- Gestione degli allegati PMA e delle notifiche a 30 giorni
- Presentazioni annuali di rapporti periodici
- Simulazioni di audit e formazione su 21 CFR 820

- Esperienza nella gestione di molteplici sottomissioni FDA PMA per diverse categorie di dispositivi.
- Team di esperti per la domanda di approvazione pre-commercializzazione della FDA secondo i requisiti normativi.
- Supporto aggiuntivo per la gestione delle domande relative alla PMA.
- Presentazione puntuale dei documenti
- Aggiornato con i nuovi emendamenti della US FDA
