

3 min di lettura
Intraprendere una sperimentazione clinica è un passo fondamentale nello sviluppo di un farmaco o di un dispositivo medico. Una Domanda di Autorizzazione per Sperimentazioni Cliniche (CTA) è richiesta alle aziende farmaceutiche o agli sponsor per ottenere l'approvazione delle autorità regolatorie per iniziare studi sull'uomo. Poiché la ricerca clinica diventa sempre più globale e complessa, comprendere le pratiche internazionali in evoluzione, i passaggi critici per la sottomissione e i vantaggi offerti da un partner o più partner per gli Affari Regolatori è cruciale sia per gli sponsor che per le organizzazioni di ricerca.
Domanda di Autorizzazione per Sperimentazioni Cliniche (CTA) e le sue Sezioni:
Una Domanda di Autorizzazione per Sperimentazioni Cliniche (CTA) è il dossier regolatorio che gli sponsor devono presentare alle autorità regolatorie nazionali (NRA) prima di iniziare uno studio clinico che coinvolga partecipanti umani. Le CTA sono obbligatorie per la maggior parte delle sperimentazioni interventistiche su farmaci, dispositivi o prodotti biologici. Esse garantiscono che la sperimentazione sia scientificamente valida, eticamente appropriata e allineata con le linee guida locali e internazionali, come gli standard di Buona Pratica Clinica (GCP) dell'International Council for Harmonization (ICH) e i requisiti nazionali.
Le sezioni chiave di una tipica CTA includono (ma non si limitano a):
- Lettera di accompagnamento e moduli di domanda
- Protocollo di studio clinico e brochure dello sperimentatore
- Dossier del prodotto e prove di Buone Pratiche di Fabbricazione (GMP)
- Approvazioni etiche e del Comitato Etico (IRB)
- Documentazione assicurativa e di protezione dei soggetti
- Piano di monitoraggio della sicurezza dei dati e dichiarazioni finanziarie
Pratiche Globali nelle Sottomissioni CTA
I processi e i requisiti delle CTA differiscono tra le regioni, ma stanno emergendo diversi principi comuni:
1. Armonizzazione di Formati e Standard
- Molti paesi utilizzano il Documento Tecnico Comune (CTD) dell'ICH, semplificando le sottomissioni di studi multinazionali. La sua struttura modulare supporta la revisione parallela e semplifica gli aggiornamenti.
- Gli adattamenti locali sono comuni, come nelle regioni dell'Asia-Pacifico e del Pacifico Occidentale, dove i paesi spesso combinano il CTD con moduli nazionali, feedback di revisione, esigenze linguistiche o studi ponte.
2. Consulenza Regolatoria
- Un coinvolgimento precoce con le agenzie, come la FDA US, le autorità nazionali dell'UE e le agenzie in Giappone, Cina e nei principali mercati emergenti, è incoraggiato, specialmente per gli studi clinici globali e multi-regionali (MRCT). Gli incontri di consulenza scientifica riducono i futuri rifiuti e guidano l'ottimizzazione del protocollo.
3. Percorsi Collaborativi e Accelerati
- Le agenzie accettano sempre più valutazioni "per affidamento" o collaborative, dove sfruttano le revisioni o le approvazioni di autorità regolatorie rigorose, aumentando la velocità e la coerenza.
- Opzioni di revisione accelerata o rapida possono essere disponibili, specialmente durante emergenze sanitarie o per terapie innovative.
4. Integrazione della Revisione Etica
- Nell'UE e in molte altre regioni, le revisioni etiche e regolatorie possono avvenire in parallelo o tramite piattaforme coordinate per evitare presentazioni separate e ridurre i tempi di avvio.
5. Trasparenza e Pubblicazione
- È ormai prassi standard registrare gli studi in database riconosciuti prima dell'approvazione, e molti paesi rendono pubblici gli stati di approvazione delle CTA, contribuendo alla trasparenza globale e alle migliori pratiche.
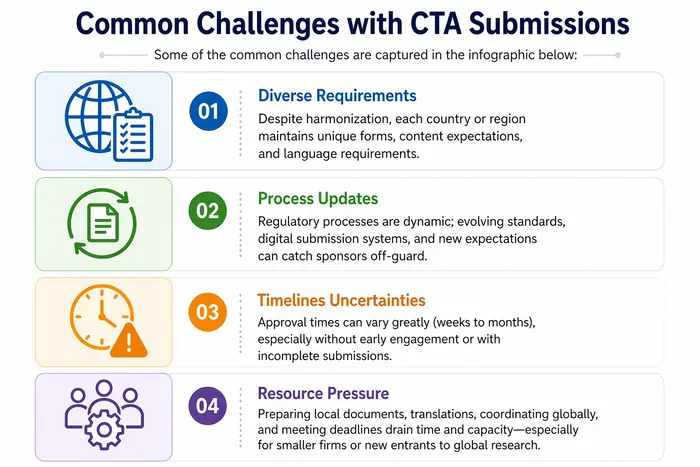
Come può aiutare un partner come Freyr?
Un partner affidabile per gli Affari Regolatori (RA) come Freyr fornisce supporto continuo e mitigazione del rischio durante l'intero processo CTA. Ecco perché coinvolgere un tale esperto prepara gli sponsor al successo:
1. Global Regulatory intelligence aggiornata
- Monitoriamo costantemente i cambiamenti regolatori, le interpretazioni regionali e le pratiche di presentazione in tutto il mondo. Ciò garantisce che ogni CTA sia strutturata in base ai requisiti più attuali, riducendo al minimo domande, rifiuti e costosi ritardi negli studi.
2. Preparazione e Revisione del Dossier
- Gli specialisti di Freyr sviluppano e compilano le CTA utilizzando i più recenti formati ICH CTD e modelli nazionali personalizzati. Coordinano le traduzioni dei documenti, organizzano le risposte ai dossier e assicurano qualità uniforme e conformità GCP per ogni presentazione.
3. Consulenza Regolatoria e Collegamento con le Agenzie
- Freyr può rappresentare gli sponsor negli incontri di consulenza scientifica pre-CTA, gestire le domande delle agenzie dopo la presentazione e supportare le comunicazioni sia con le autorità che con i Comitati Etici.
4. Gestione Progetti Globale
- Con team profondamente multiculturali e strumenti di gestione progetti robusti, Freyr coordina le presentazioni simultanee, monitora i progressi paese per paese e promuove l'allineamento per gli MRCT (Studi Clinici Multi-Regionali).
5. Strategia di Affidamento e Revisione Accelerata
- Freyr aiuta gli sponsor a sfruttare i quadri di affidamento, a preparare dossier abbreviati per revisioni esonerate o accelerate e ad allineare i dati di sottomissione per la massima accettazione regolatoria.
6. Prontezza per Audit e Controllo delle Modifiche
- Il partner garantisce che la documentazione e le procedure degli sponsor soddisfino gli standard di audit, facilitando una rapida approvazione regolatoria e ispezioni senza intoppi.
Conclusione
La sottomissione di una CTA è un passaggio complesso ma essenziale per la ricerca clinica, con requisiti globali e specifici per regione. Le tendenze globali includono l'armonizzazione del CTD, la revisione etica e regolatoria parallela, i meccanismi di affidamento e le nuove norme sulla trasparenza. Gli sponsor affrontano sfide significative nel soddisfare requisiti diversi, nel tenere il passo con i cambiamenti regolatori e nel gestire le tempistiche globali. In tale scenario, un partner per gli Affari Regolatori come Freyr fornisce competenze End-to-End in intelligence regolatoria, preparazione dei dossier, interazioni con le agenzie, percorsi accelerati e prontezza per l'audit, aumentando sostanzialmente le probabilità di avvii tempestivi e di successo delle sperimentazioni cliniche.









