
4 min de lecture
Pour la plupart des équipes chargées des affaires réglementaires, le renouvellement d'un enregistrement n'est pas simplement une étape administrative de plus. C'est le moment où des années de dossiers qualité, de données post-commercialisation et d'historique des modifications sont examinées de près par les autorités réglementaires.
Pourtant, les renouvellements sont souvent abordés de manière réactive.
Les fichiers sont rassemblés à la dernière minute. Les données du système de gestion des commandes (PMS) sont dispersées dans différents dossiers. Les certificats sont répertoriés dans des tableurs. Et soudain, ce qui aurait dû être un processus structuré se transforme en une course contre la montre.
Les renouvellements de dispositifs médicaux n'échouent pas parce que les équipes ne font pas d'efforts. Ils échouent parce que la planification du renouvellement commence trop près de la date d'expiration.
Une stratégie de renouvellement solide change la donne. Elle considère le renouvellement comme une activité qui s'inscrit dans un cycle de vie continu, et non comme une simple formalité de dernière minute.
Voyons ce que cela signifie concrètement pour RA .
Pourquoi le renouvellement des dispositifs médicaux nécessite une stratégie
La plupart des enregistrements de dispositifs sont valables pendant 3 à 5 ans, selon le marché. Certaines régions ont recours à des renouvellements annuels, tandis que d'autres appliquent des modèles de maintien de licence avec des paiements périodiques. Quelle que soit la structure, une fois la licence expirée, le dispositif ne peut plus être légalement fabriqué, importé ou distribué.
Les demandes de renouvellement présentent un risque élevé, car les autorités procèdent à une nouvelle évaluation :
- Performance après la mise sur le marché
- Tendances en matière de plaintes et événements indésirables
- Modifications apportées au système de fabrication et de qualité
- Données cliniques et de sécurité
- Historique de conformité réglementaire
Dans de nombreuses régions, notamment en Inde et dans l'Union européenne, les autorités de réglementation exigent la présentation de données PMS consolidées, de preuves des mesures correctives et préventives (CAPA) ainsi que d'une documentation technique mise à jour dans le cadre de l'examen de renouvellement.
C'est pourquoi le renouvellement doit être considéré comme une discipline opérationnelle qui s'applique tout au long du cycle de vie du produit.
Pas comme une tâche soumise à des délais.
Commencez par un calendrier réglementaire : votre première ligne de défense
L'une des principales causes d'échec des renouvellements réside dans un suivi fragmenté. Des marchés différents. Des calendriers différents. Des exigences différentes.
Un calendrier réglementaire centralisé devrait inclure :
- Numéros de certificats et dates d'expiration par pays
- Périodes de dépôt des demandes de renouvellement
- Délais de paiement des honoraires de conservation
- Validité des certificats ISO 13485 CE
- Étapes clés internes (12 mois, 6 mois, 3 mois avant le renouvellement)
D'un RA pratique RA , la planification devrait débuter au moins 12 mois avant l'expiration.
De nombreuses équipes ont désormais recours à des plateformes RIMS unifiées pour disposer d'une vue d'ensemble du statut des enregistrements sur l'ensemble des marchés, ce qui leur permet d'éviter de dépendre des tableurs tout en conservant une visibilité sur les délais de renouvellement et en garantissant leur suivi.
freya fusion la première solution de ce type à permettre aux organisations de disposer d'une source unique et fiable pour toutes leurs données relatives à la conformité.
Les documents essentiels dont vous aurez toujours besoin
Les documents de renouvellement sont très similaires à ceux de l'enregistrement initial, mais mettent davantage l'accent sur la continuité et les performances.
La plupart des autorités s'attendent à ce que :
- Lettre de motivation indiquant clairement l'intention de renouveler
- Déclaration de conformité continue (fabrication, conception, propriété)
- Mise à jour du fichier de référence des dispositifs et du fichier de référence des sites (le cas échéant)
- ISO 13485 en cours de validité
- Certificats CE ou agréments en matière d'assurance qualité, le cas échéant
- Licences de fabrication et justificatifs de conformité aux BPF
- Déclaration de conformité
- Historique de la correspondance réglementaire
Toute modification survenant pendant la période de validité doit être signalée par les voies prévues à cet effet après l'autorisation. Les modifications non déclarées constituent l'un des moyens les plus rapides de susciter des questions de la part des autorités réglementaires.
C'est là que la gestion structurée des documents prend tout son sens. RA tirent profit du fait que toutes les soumissions, révisions et validations soient regroupées dans un environnement contrôlé, plutôt que dispersées entre différents disques durs et boîtes de réception.
La surveillance post-commercialisation est au cœur du renouvellement
S'il y a bien un domaine que les autorités de régulation doivent examiner de près lors du renouvellement, c'est celui de la surveillance post-commercialisation.
Les autorités attendent des preuves démontrant que le dispositif a continué à fonctionner de manière sûre et efficace dans des conditions réelles d'utilisation.
En général, cela comprend :
- Données de vente par année et par modèle d'appareil
- Registres des plaintes et rapports d'événements indésirables
- Analyse des causes profondes pour chaque problème
- Dossiers CAPA accompagnés de preuves de clôture
- Historique des rappels (au niveau mondial et par pays)
- Analyse des tendances sur toute la période de validité
Les dispositifs présentant un risque plus élevé peuvent également nécessiter :
- Rapports PMS
- Mises à jour du rapport d'évaluation clinique
- PMCF
- Rapports périodiques sur la sécurité
La qualité des résultats en matière de renouvellement dépend de la régularité avec laquelle ces données sont collectées au fil du temps, et non de la qualité de leur compilation finale.
De nombreuses RA intègrent désormais le suivi des PMS directement dans leurs processus réglementaires à l'aide de plateformes unifiées telles que freya fusion, ce qui permet de relier entre eux les rapports d'incidents, les mesures correctives et préventives (CAPA) et les enregistrements, pour une traçabilité facilitée lors de la préparation des renouvellements.
Adaptez votre système qualité pour être prêt pour le renouvellement
Votre système de gestion de la qualité n'est pas distinct du processus de renouvellement. Il en constitue le fondement.
Les autorités de régulation recherchent des éléments probants dans des domaines clés tels que :
- Contrôle des documents et historique des versions
- Gestion du changement dans les domaines de la fabrication, de l'étiquetage et du conditionnement
- Qualification et audits des fournisseurs
- Traitement des réclamations et analyse des tendances
- Efficacité du CAPA
- Audits internes
- Dossiers de formation et de compétences
Une approche pratique consiste à réaliser une analyse des lacunes avant le renouvellement, environ six mois avant le dépôt de la demande :
- Vérifier que les certificats resteront valides après examen
- Vérifier que les données du système de gestion des stocks (PMS) sont complètes
- Vérifier les fermetures CAPA
- Consulter l'historique des modifications
- Veiller à ce que les dossiers de formation soient à jour
Cet audit proactif permet souvent d'éviter les mesures correctives de dernière minute.
Anticipez dès le départ la complexité liée à la présence sur plusieurs marchés
Les renouvellements se font rarement de manière isolée. Les portefeuilles internationaux impliquent différents agents, distributeurs, organismes notifiés et autorités.
La coordination comprend généralement :
- Fabricant fournissant des dossiers techniques et des données PMS
- Agents locaux chargés de la gestion des demandes et des licences
- Distributeurs partageant leurs données relatives aux ventes et aux réclamations
- Organismes notifiés délivrant des certificats d'audit
En l'absence de responsabilités clairement définies et de délais précis, les retards s'accumulent.
Un système centralisé permettant de suivre les exigences propres à chaque juridiction, l'état d'avancement des dépôts et les communications réglementaires peut réduire considérablement les frictions. C'est là que freya fusion RA en regroupant les enregistrements, les documents et la collaboration entre les parties prenantes au sein d'un espace de travail réglementaire unifié.
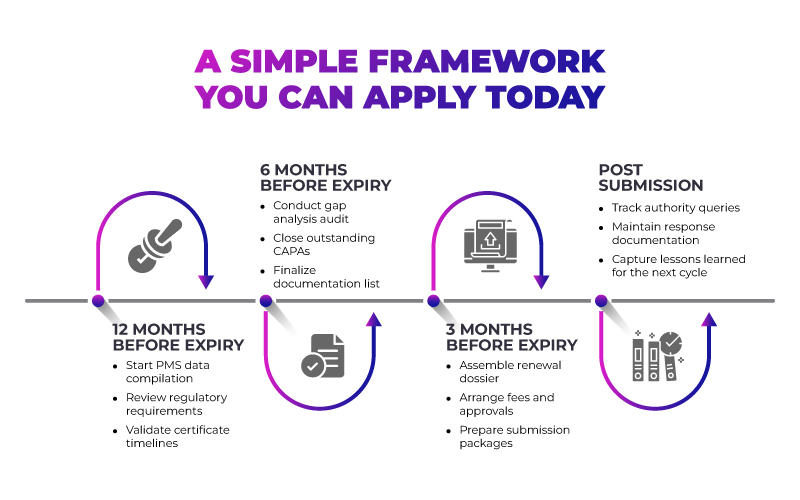
Cette approche par étapes fait du renouvellement un processus prévisible plutôt qu'une crise récurrente.
Le renouvellement, c'est une question de continuité, et pas seulement de conformité
Le renouvellement de l'enregistrement des dispositifs médicaux vise avant tout à garantir un accès ininterrompu au marché. Lorsqu'il est géré de manière stratégique, il renforce les systèmes qualité, améliore le respect des obligations liées au suivi post-commercialisation et renforce la confiance des autorités réglementaires.
RA fournissent déjà un travail acharné au quotidien. Le défi consiste à donner à ce travail une structure, une visibilité et une continuité.
Ici, des plateformes telles que freya fusion entrent en jeu. En centralisant les enregistrements, les documents, les systèmes de gestion des dossiers (PMS) et la collaboration, elles aident RA à passer d'un renouvellement réactif à une gestion proactive du cycle de vie réglementaire.
Prêt à simplifier le renouvellement de vos dispositifs médicaux ?
Si les renouvellements vous semblent encore dispersés, fastidieux ou stressants, il est peut-être temps de repenser l'organisation de vos opérations réglementaires.
Découvrez comment freya fusion, grâce à ses fonctionnalités axées sur l'IA, aide RA des dispositifs médicaux en leur offrant un suivi centralisé des enregistrements, un contrôle des documents et une visibilité sur le cycle de vie, le tout conçu pour les professionnels de la réglementation et non pour les équipes informatiques.
Réservez une démonstration dès aujourd'hui et faites le premier pas vers un regain de confiance, une conformité permanente et une continuité des activités plus fluide.








