

4 min de lecture
Avec la mise en œuvre par la Food and Drug Administration des US (USFDA) de la règle finale concernant les réglementations du système de gestion de la qualité (QMSR) en 2024, les fabricants de dispositifs médicaux doivent adopter les amendements pour commercialiser et distribuer leurs dispositifs sur le marché des USA.
Cette règle actualise la réglementation sur les systèmes qualité (QSR) USFDAen l'alignant sur la norme ISO 13485:2016, qui est la norme internationale relative aux systèmes de gestion de la qualité des dispositifs médicaux. Les fabricants de dispositifs médicaux disposent d'une période de transition de deux ans pour se mettre en conformité, ce qui oblige les organisations à s'adapter aux nouvelles exigences afin d'éviter toute non-conformité lors des inspections.
Qu'est-ce que le QMSR ?
FDA constitue une approche simplifiée des exigences en matière de système de gestion de la qualité (QMS) et fait suite à la structure antérieure du QSR. Cette harmonisation est essentielle, car elle facilite la mise en conformité à l'échelle mondiale pour les fabricants, en particulier ceux qui opèrent à l'international. Elle permettra aux entreprises de satisfaire aux exigences réglementaires tant aux US sur d'autres marchés de manière beaucoup plus cohérente.
Le QMSR exige des améliorations en matière de gestion des risques, de conception des dispositifs et de surveillance après commercialisation. Ce rôle peut ajouter de la complexité, mais il permet également aux fabricants de normaliser leurs procédures qualité, ce qui, à son tour, améliore la sécurité des dispositifs et la documentation, un élément vital lors des inspections de l'USFDA.
Principaux changements du QMSR
- Harmonisation avec ISO 13485: il s'agit de l'étape la plus cruciale, qui permet aux fabricants de dispositifs médicaux d'adopter des normes reconnues à l'échelle internationale. USFDA que de nombreuses entreprises du secteur des dispositifs médicaux se conformaient déjà à ISO 13485, ce qui évite les efforts redondants.
- L'approche de gestion des risques met l'accent sur la gestion des risques tout au long du cycle de vie du dispositif médical. Les fabricants de dispositifs médicaux doivent faire preuve d'une gestion des risques, d'un contrôle des risques et d'une gestion des risques efficaces.
- Conception et contrôles des dispositifs : Dans le cadre du QMSR, les contrôles de conception ont été étendus pour garantir que les fabricants de dispositifs médicaux tiennent pleinement compte des besoins des utilisateurs, de la sécurité du dispositif et des critères de performance, ce qui est un domaine d'attention particulier lors des inspections de l'USFDA.
- Surveillance après commercialisation : Les entreprises doivent améliorer le système de surveillance après commercialisation. Cela implique que les fabricants recueillent des informations sur la sécurité et l'efficacité du dispositif, ce qui aide à identifier et à résoudre rapidement les problèmes.
- Documentation et tenue des registres : C'est la règle finale qui met l'accent sur la documentation. Une tenue des registres rigoureuse / appropriée est essentielle lors de l'inspection.
Étapes pour se préparer à l'inspection de la US Food and Drug Administration :
Avec l'inspection qui dure jusqu'en 2026, les entreprises ont deux ans pour aligner leurs systèmes qualité sur le QMSR. Cependant, attendre la dernière minute peut être risqué.
Étapes pour l'inspection de l'USFDA dans le cadre du QMSR pour les industries dont le QMS actuel est basé sur le QSR :
- Réaliser une analyse des écarts : C'est la première étape où le système qualité actuel s'écarte des nouvelles exigences du QMSR. Une analyse approfondie des écarts permettra d'identifier les domaines nécessitant des mises à jour, tels que la gestion des risques, la surveillance après commercialisation et les contrôles de conception.
- Mettre à jour la procédure de gestion des risques : assurez-vous que les activités de gestion des risques sont intégrées à l'ensemble du cycle de vie de votre produit, de la conception à la surveillance après commercialisation.
- Revoir le contrôle de la conception : Les fabricants doivent s'assurer que le processus de conception est robuste et bien documenté. Valider que le processus de conception est robuste, bien documenté et entièrement intégré à votre système de gestion de la qualité.
- Améliorer la surveillance après commercialisation : Mettre en place des systèmes pour surveiller la performance du dispositif après sa mise sur le marché. Cela pourrait impliquer la mise en place de mécanismes de retour client, la collecte de preuves de données cliniques et leur suivi rigoureux.
- Formation et documentation : Former le personnel aux nouvelles exigences, en particulier ceux qui sont impliqués dans la gestion de la qualité et la conformité réglementaire. Cela garantit que tout le processus de documentation est aligné sur les attentes du QMSR.
- Certification par un organisme tiers : si votre entreprise n'est pas ISO 13485 , c'est peut-être le moment d'y songer. Obtenir ISO 13485 peut vous donner une longueur d'avance pour répondre aux exigences de USFDA renforcer votre crédibilité sur les marchés mondiaux.
Gérer la période de transition de deux ans
Pour utiliser efficacement la période de transition et assurer la conformité avec le Règlement sur le Système de Gestion de la Qualité (QMSR) de l'USFDA, les fabricants devraient adopter une approche proactive. Voici une feuille de route suggérée :
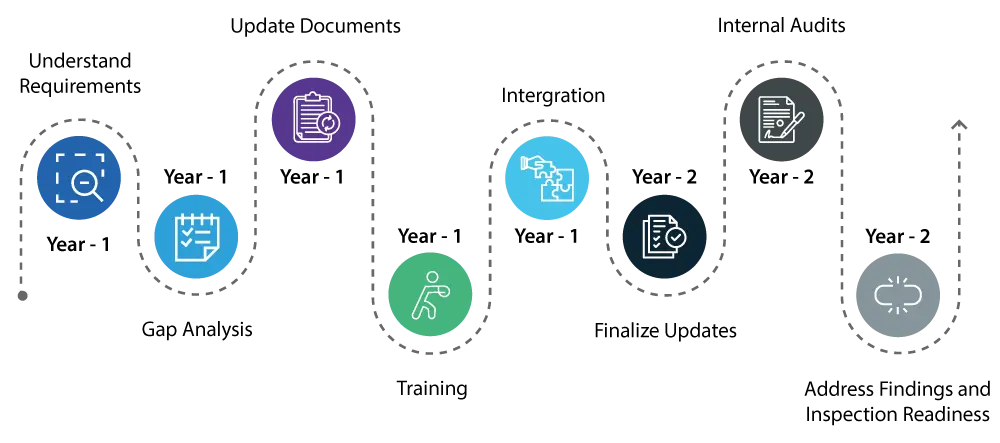
Année 1 :
- Comprendre l'exigence : Commencez par bien comprendre les changements introduits par le QMSR. Cela implique un examen détaillé des nouvelles exigences et de la manière dont elles diffèrent du règlement existant sur le système qualité (QS).
- Identifiez les lacunes : Menez une analyse complète des écarts pour identifier les aspects de votre système qualité actuel qui nécessitent des mises à jour afin de respecter les nouvelles normes QMSR.
- Mise à jour / Remédiation des documents : Initier les mises à jour nécessaires de votre système qualité, en se concentrant sur des domaines tels que la gestion des risques et les contrôles de conception, qui sont des composants essentiels du QMSR.
- Formation : Commencez à former votre personnel aux nouvelles réglementations afin de s'assurer que toutes les personnes concernées sont informées des changements et comprennent leurs rôles dans le maintien de la conformité.
- Intégration : Commencez à intégrer les nouvelles exigences QMSR dans vos opérations quotidiennes pour faciliter la transition.
Année 2 :
- Poursuivez la mise en œuvre des changements apportés à votre système qualité, en veillant à ce que toutes les mises à jour soient entièrement intégrées et opérationnelles.
- Mener des audits internes approfondis pour vérifier que les mises à jour sont efficaces et que votre système qualité est entièrement aligné sur les exigences du QMSR.
- Traiter rapidement toutes les constatations des audits internes afin de garantir que tous les aspects de votre système qualité sont conformes.
- D'ici la fin de la deuxième année, votre système qualité devrait être entièrement conforme au QMSR, et vous devriez être préparé aux inspections de l'USFDA avec la certitude qu'il n'y aura pas de problèmes majeurs.
En suivant cette feuille de route, les fabricants peuvent non seulement satisfaire aux exigences de l'USFDA, mais aussi établir un système qualité robuste, efficace, standardisé et reconnu mondialement. Cette approche proactive contribuera à assurer une transition en douceur vers les nouvelles réglementations et à maintenir les normes les plus élevées de qualité et de sécurité pour les dispositifs médicaux.
Conclusion : Adopter des mesures proactives
Se préparer aux USFDA dans le cadre de la nouvelle réglementation QMSR ne consiste pas seulement à éviter les sanctions, mais aussi à améliorer la sécurité et l'efficacité des dispositifs médicaux. En s'alignant sur ISO 13485, USFDA des exigences plus élevées, mais offre également une voie vers une conformité mondiale simplifiée. Les fabricants qui s'adaptent rapidement doivent se concentrer sur des domaines clés tels que la gestion des risques et la surveillance post-commercialisation, et s'assurer que leurs systèmes qualité sont à jour et solides, ce qui leur permettra non seulement de répondre aux attentes réglementaires, mais aussi de renforcer leur avantage concurrentiel sur le marché.
