
5 min de lecture
Le Programme d'audit unique des dispositifs médicaux (MDSAP) permet à un organisme d'audit agréé (AO) de réaliser un audit unique du système de gestion de la qualité (SGQ) d'un fabricant de dispositifs médicaux. Il couvre les exigences réglementaires applicables dans cinq pays, à savoir le Brésil (ANVISA), les États-Unis (FDA), le Japon (PMDA), le Canada (Santé Canada) et l'Australie (TGA). Outre les autorités réglementaires participantes, plusieurs autres partenaires internationaux (les observateurs officiels et les membres affiliés) sont impliqués dans le MDSAP.
La certification MDSAP est obligatoire pour les dispositifs de classe II, III et IV selon Santé Canada, mais elle est volontaire pour les quatre autres pays. Elle a favorisé la transparence et l'harmonisation réglementaire entre les autorités participantes et a minimisé le besoin d'audits multiples, permettant ainsi d'économiser du temps et des ressources pour les fabricants de dispositifs médicaux. Pour vous donner une meilleure perspective sur le programme MDSAP, nous avons tenté de répondre aux quinze (15) questions les plus fréquemment posées.
- Pourquoi MDSAP a-t-il alors qu'il existe déjà une ISO 13485 reconnue mondialement ?
Le MDSAP a été développé pour alléger le fardeau des audits réglementaires pour les fabricants de dispositifs médicaux et pour promouvoir une meilleure harmonisation des approches réglementaires et des exigences techniques basées sur les normes internationales et les meilleures pratiques. Il vise à apporter cohérence, prévisibilité et transparence aux programmes réglementaires en standardisant les procédures et les pratiques des régulateurs et des organismes d'audit tiers.
L'audit repose sur les exigences du système de gestion de la qualité (SGQ) prévues par ISO 13485 sur les exigences réglementaires du pays participant dans lequel les dispositifs médicaux seront commercialisés.
- Quels sont les critères d'éligibilité pour être soumis à un audit MDSAP ?
Tout fabricant de dispositifs médicaux souhaitant commercialiser son dispositif dans les pays participants peut se soumettre à un audit MDSAP. Cependant, chaque autorité réglementaire peut établir des critères d'exclusion pour certaines conditions si nécessaire.
Par exemple, au Japon, les exceptions d'éligibilité sont :
- Un Site de Fabrication Enregistré (RMS) qui fabrique des dispositifs médicaux à partir de tissus humains/animaux
- Un RMS qui fabrique des DIV radioactifs, et
- La désignation d'un titulaire de l'autorisation de mise sur le marché (MAH)
- L'audit MDSAP inclut-il les produits combinés ?
Les dispositifs médicaux qui contiennent des médicaments (substances médicinales) ou des produits biologiques (par exemple, des matériaux d'origine animale rendus non viables, ou des tissus, des cellules, ou des substances d'origine microbienne ou recombinante, du sang humain ou des extraits de sang humain ou des produits sanguins, etc.) sont considérés comme des produits combinés et peuvent être inclus dans le champ d'application d'un audit MDSAP.
Cependant, en raison des différences dans la manière dont ces produits sont réglementés dans les juridictions des autorités réglementaires participantes, les rapports d'audit MDSAP et les documents de certification peuvent ne pas être considérés comme une alternative aux exigences d'inspection et d'évaluation dans certaines juridictions.
Australie - Les produits combinés sont soumis à un examen hors site de la TGA dans le cadre de l'évaluation de la conformité australienne. Cependant, un audit MDSAP efficace peut réduire le nombre d'inspections pour ces dispositifs.
Brésil, Japon- Les produits combinés considérés comme des dispositifs médicaux sont inclus dans le MDSAP, car il n'y a pas d'exigences spécifiques concernant le SMQ.
Canada- Le modèle MDSAP couvre les exigences du SMQ pour les produits combinés considérés comme des dispositifs médicaux.
États-Unis – MDSAP ne sont pas considérés comme des alternatives aux FDA pour les produits combinés.
- Puis-je sélectionner le pays concerné par l'audit MDSAP ?
Oui, l'audit est réalisé conformément au périmètre déclaré dans la demande de services de certification. Les fabricants de dispositifs médicaux sont tenus de se conformer aux réglementations uniquement dans les juridictions où leurs produits doivent être commercialisés.
- Je suis un fabricant de dispositifs médicaux des US, ayant l'intention de commercialiser mon dispositif uniquement au Japon. Je suis sur le point de subir un audit MDSAP. Dois-je également me conformer aux exigences d'autres pays ?
Non, les fabricants de dispositifs médicaux ne sont tenus de se conformer aux ISO 13485 de ISO 13485 et à la réglementation en vigueur que dans les pays où leurs produits sont destinés à être commercialisés.
- Mon organisme d'audit (AO) et mon organisme notifié européen sont les mêmes. Puis-je être audité pour les deux en même temps ?
Si votre organisme d'évaluation de la conformité (AO) et votre organisme notifié européen sont identiques, l'évaluation de la conformité peut être réalisée après MDSAP , et non simultanément. Les organismes notifiés européens ont un statut d'observateurs dans le cadre MDSAP, et l'évaluation de la conformité est menée conformément au règlement EU MDR . Dans le cadre MDSAP, l'évaluation est réalisée conformément aux exigences de ISO 13485 aux exigences réglementaires des pays participants concernés.
- Quelle est la différence entre les évaluations de stade I et II ?
Le processus d'audit initial du MDSAP comporte deux étapes. L'audit initial, également appelé audit de certification initiale, se compose d'audits de phase I et de phase II.
L'audit de phase I comprend l'examen des documents et l'évaluation de l'aptitude du fabricant de dispositifs médicaux à subir un audit de phase II.
L'audit de phase II est réalisé afin de vérifier si toutes les exigences applicables de ISO 13485 les autres exigences réglementaires de l'autorité de réglementation concernée sont mises en œuvre.
- Combien d'auditeurs puis-je attendre pour un audit MDSAP ?
La détermination de la durée de l'audit précise comment établir la durée d'un audit sur site en jours-homme. L'AO décide du nombre d'auditeurs qui composeront l'équipe d'audit. Par exemple, un audit de (06) jours-homme peut être réalisé en trois (03) jours par une équipe de deux (02) auditeurs.
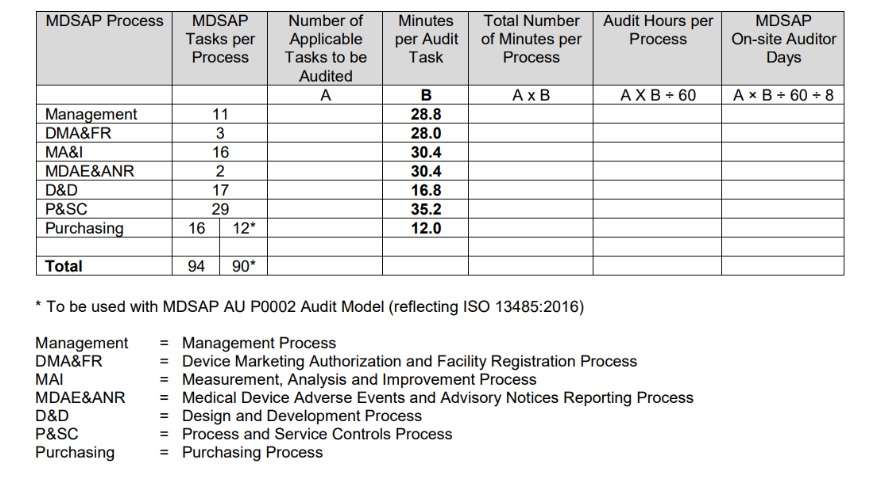
- Comment l'audit MDSAP est-il planifié ?
La procédure de détermination de la durée de l'audit, publiée par la FDA, résume dans le tableau ci-dessous le processus permettant de déterminer la durée de l'audit.
Le calcul de la durée de l'audit repose principalement sur le nombre de tâches d'audit applicables associées au type d'audit à réaliser et aux activités spécifiques de l'organisation à auditer.
Pour des informations détaillées à ce sujet, vous pouvez vous référer à MDSAP P0008007.
- Existe-t-il un guide ou une liste de contrôle que je peux consulter pour assurer la conformité à un audit MDSAP ?
Oui, vous pouvez consulter le document « MDSAP Approach ». Il s'agit d'un guide bien structuré publié par la USFDA renvoie à des sections spécifiques de ISO 13485:2016 et aux réglementations pertinentes émises par la TGA australienne, ANVISA brésilienne, Santé Canada,PMDA japonais et laFDA US .
- Quel est le rôle d'un observateur lors d'un audit MDSAP ?
Un observateur MDSAP est une Autorité Réglementaire qui est autorisée à assister aux réunions, évaluations et autres activités, mais qui n'utilise pas les livrables du MDSAP. Les observateurs sont représentés au Conseil des Autorités Réglementaires du MDSAP (RAC) par un gestionnaire de haut niveau.
- Quelles sont les prochaines étapes à suivre si j'ai obtenu une note de 4 ou plus ?
Le système de notation est appliqué aux non-conformités observées lors de l'audit par l'AO. Un score de 4 ou 5 indique un risque élevé nécessitant une intervention. Vous devez fournir un plan de remédiation pour chaque non-conformité enregistrée dans les 15 jours calendaires suivant la date d'émission du rapport de non-conformité. Le plan de remédiation doit inclure les résultats de l'enquête sur la non-conformité, ses causes et les actions correctives prévues pour éviter toute récurrence. La preuve de la mise en œuvre du plan de remédiation/d'action doit être fournie dans les trente (30) jours calendaires suivant la date de fin de l'audit.
- Y a-t-il une différence dans le processus d'approche de l'audit par un auditeur interne par rapport à un AO ?
Le MDSAP adopte une approche par processus. L'AO est susceptible d'examiner les liens et les interconnexions, tandis qu'un auditeur interne pourrait se concentrer sur un seul aspect fonctionnel à la fois. Par conséquent, l'AO pourrait identifier une non-conformité dans un domaine fonctionnel et chercher des réponses dans un autre domaine fonctionnel. Cependant, suivre l'approche par processus pourrait être perturbateur lors d'un audit interne.
- Puis-je faire appel auprès de l'AO si je peux prouver qu'une non-conformité enregistrée n'est pas valide ?
L'AO dispose d'un processus d'appel ou de litige, que vous pouvez utiliser si vous pouvez démontrer qu'une non-conformité enregistrée est invalide. Cependant, les niveaux attribués aux non-conformités ne peuvent pas être modifiés en raison d'actions correctives. Ils ne peuvent être modifiés que sur la base de preuves démontrant qu'ils n'étaient pas valides.
- Quelle est la durée de validité du certificat MDSAP ?
Les fabricants de dispositifs médicaux certifiés dans le cadre du programme MDSAP seront audités annuellement, selon un cycle de certification de trois ans. L'audit initial est un audit complet du SGQ du fabricant de dispositifs médicaux. Il est suivi d'audits de surveillance menés annuellement pendant deux (02) années consécutives. Le cycle reprend avec un audit de recertification la troisième année.
Pour en savoir plus sur nos services MDSAP, contactez Freyr dès aujourd'hui pour planifier un appel avec nos experts.









