Pourquoi la surveillance post-commercialisation est-elle essentielle dans le cycle de vie des dispositifs médicaux ?
La surveillance post-commercialisation (PMS) est un élément essentiel du cycle de vie des dispositifs médicaux, en particulier dans un contexte réglementaire en constante évolution, comme l'exigent EU MDR, le RIDM et les exigences FDA . À mesure que les dispositifs gagnent en sophistication, qu'il s'agisse de SaMD, de dispositifs portables ou de technologies connectées, une surveillance continue en conditions réelles et une détection proactive des risques sont indispensables. Les tendances du secteur, telles que les données issues du monde réel (RWE), la vigilance numérique et les résultats rapportés par les patients, soulignent la nécessité d'une évaluation continue de la sécurité, des performances et des bénéfices cliniques des dispositifs. Dans le cadre des attentes actuelles en matière de surveillance post-commercialisation, les fabricants doivent de plus en plus intégrer des rapports sur les tendances, la détection des signaux et des évaluations fondées sur les risques.

Malgré son importance, de nombreux fabricants ont du mal à répondre aux exigences du système de surveillance post-commercialisation (PMS). La fragmentation des sources de données, l'incohérence des obligations de déclaration à l'échelle mondiale, la gestion multilingue des canaux de signalement des effets indésirables et le renforcement de la surveillance réglementaire sont autant de facteurs qui compliquent les opérations. La préparation des plans PMS, des rapports PMSR, des rapports PSUR et PMCF nécessite une expertise spécialisée, une interprétation précise des données et une coordination interfonctionnelle. Des lacunes dans l'analyse des tendances, la détection des signaux, l'évaluation des risques pour la santé (HHE) ou la notification de vigilance relative aux dispositifs médicaux peuvent entraîner des constatations d'audit, des rappels potentiels ou des risques pour la continuité des produits.
Freyr relève ces défis grâce à des solutions complètes end-to-end , conçues pour répondre aux exigences réglementaires mondiales. Nos équipes allient expertise réglementaire, méthodologies structurées et assistance multilingue pour rationaliser le traitement des plaintes, la notification des incidents et la documentation relative au PMS. Fort d'une expérience éprouvée dans SaMD de classe I à III, SaMD de diagnostic in vitro (IVD) et SaMD , Freyr garantit des rapports de haute qualité, une mise en conformité rapide et une préparation optimale aux audits, ce qui fait de nous un partenaire de confiance pour les fabricants de dispositifs médicaux à la recherche d'une gestion fiable et efficace du cycle de vie du PMS.
Éléments clés de la surveillance post-commercialisation des dispositifs médicaux
Une surveillance post-commercialisation (PMS) efficace combine plusieurs activités interdépendantes qui permettent de contrôler la sécurité des dispositifs, leurs performances cliniques, l'expérience des utilisateurs et les risques émergents tout au long du cycle de vie commercial du produit. Ces éléments constituent le fondement des exigences réglementaires mondiales prévues par EU MDR et le RID EU MDR, ainsi que par les exigences FDA . Il est essentiel de bien comprendre chacun de ces éléments pour garantir la conformité, améliorer les performances des dispositifs dans la pratique et prévenir de manière proactive les problèmes de sécurité.
![]()
Gestion des plaintes et des événements indésirables
Gestion structurée de la réception, de l'examen et de l'analyse des tendances des plaintes afin de détecter rapidement les signaux de sécurité, d'assurer une remontée hiérarchique en temps opportun et de garantir le respect, à l'échelle mondiale, des exigences FDA, EU MDR et des obligations régionales en matière de notification de vigilance.![]()
Vigilance et signalement des effets indésirables
Identification, évaluation et notification en temps opportun des événements indésirables et des incidents graves aux autorités réglementaires mondiales, afin d'assurer une surveillance continue de la sécurité et la conformité aux exigences EU MDR prévues par le règlement FDA et le EU MDR .![]()
Rappels, corrections et suppressions
End-to-end des mesures correctives sur le terrain, y compris l'évaluation des risques, l'évaluation des risques pour la santé (HHE), les notifications réglementaires, la communication et les contrôles d'efficacité, afin de garantir la sécurité des patients et de préserver la crédibilité sur le marché.![]()
s sur le régime PMS (PMSP)
Un plan structuré de surveillance post-commercialisation définissant les responsabilités, les sources de données, les processus et les critères d'évaluation afin d'assurer un suivi cohérent et proactif des performances du dispositif tout au long de son cycle de vie.![]()
PMSR, PSUR
etPMCF
Rapports exigés par la réglementation qui résument les données post-commercialisation, notamment le rapport de surveillance post-commercialisation (PMSR), les mises à jour du rapport bénéfice-risque et les activités de suivi clinique visant à démontrer la sécurité et les performances continues du dispositif.![]()
Rapports sur les tendances et données issues de la pratique clinique
Analyse des tendances en matière de réclamations et des données de performance réelles afin d'identifier les risques émergents, de soutenir les mesures préventives et d'améliorer la fiabilité des produits grâce à des pratiques structurées de reporting des tendances.






Services de surveillance après commercialisation de Freyr
Prenez rendez-vous avec nos experts dès aujourd'hui
Coup de projecteur sur les réussites : des résultats concrets, des témoignages authentiques
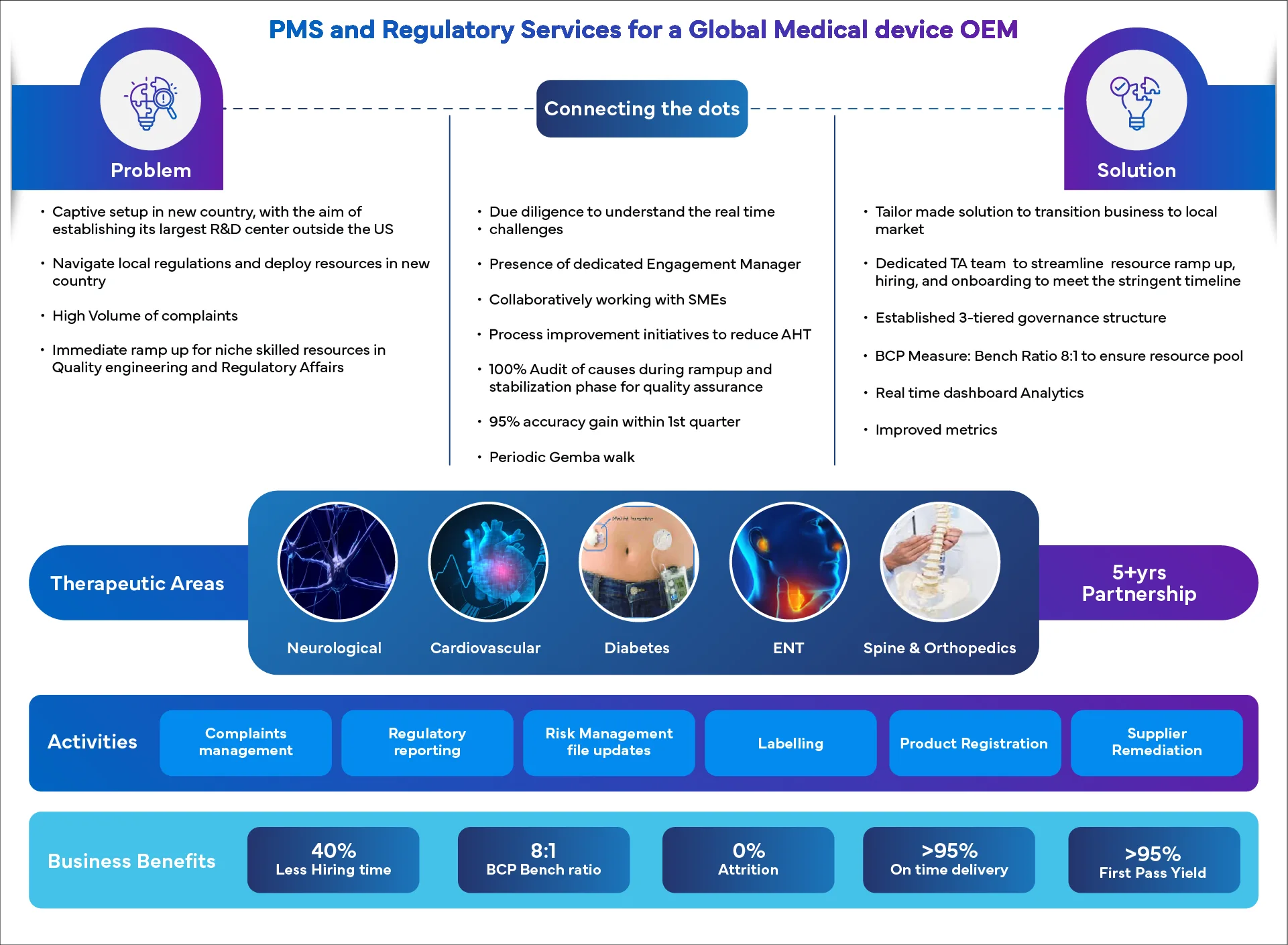
Pourquoi collaborer avec Freyr ?
- Une vaste expérience des cadres réglementaires de FDA, EU MDR, du RID et du PMS de la région APAC garantit une conformité cohérente et prête pour les inspections, ainsi qu'une forte harmonisation réglementaire sur l'ensemble des marchés mondiaux.
- End-to-end couvrant la gestion des plaintes, les rapports de vigilance, les rapports PMSR, les rapports PSUR et PMCF une gestion précise et efficace de l'ensemble du cycle de vie post-commercialisation.
- Les analyses basées sur l'IA, l'identification automatisée des tendances et les informations issues de données concrètes permettent de détecter rapidement les risques émergents, facilitent la gestion des mesures correctives et préventives (CAPA) et favorisent la prise de décisions proactives en matière de sécurité.
- Des experts maîtrisant les langues locales optimisent le tri, la documentation et le traitement des réclamations, garantissant ainsi un fonctionnement fluide et précis du système de gestion des réclamations (PMS) dans toutes les régions.
- Une expérience éprouvée dans l'accompagnement des fabricants lors des audits FDA, EU MDR et des organismes notifiés, se traduisant par une réduction des non-conformités, une amélioration des résultats en matière de qualité et une plus grande assurance quant à la conformité.
Foire aux questions
01. Qu'est-ce que la surveillance post-commercialisation (PMS) des dispositifs médicaux et pourquoi est-elle importante ?
La surveillance post-commercialisation est un processus continu et systématique visant à contrôler la sécurité, les performances et l'efficacité en conditions réelles des dispositifs médicaux après leur mise sur le marché. Elle est essentielle car elle permet aux fabricants de détecter les risques émergents, de valider les bénéfices cliniques au fil du temps, de se conformer à une réglementation mondiale en constante évolution et de prendre des décisions fondées sur des données probantes afin d'améliorer la qualité des produits et la sécurité des patients.
02. Quelles sont les principales exigences relatives aux systèmes de gestion de la qualité (PMS) prévues par EU MDR le RDI EU MDR ?
EU MDR IVDR EU MDR imposent aux fabricants de mettre en place un plan de surveillance post-commercialisation (PMS), de produire régulièrement des rapports de surveillance post-commercialisation (PSUR), de mener PMCF lorsque cela s'avère nécessaire, et de mettre en place des systèmes de notification de vigilance et d'analyse des tendances. Ces règlements mettent l'accent sur la collecte proactive de données, l'intégration des données cliniques et l'évaluation continue du rapport bénéfice/risque tout au long du cycle de vie du dispositif.
03. Comment le PMS s'intègre-t-il à la gestion des risques et à la norme ISO 14971 ?
Le système PMS intègre directement des informations concrètes dans le cadre de gestion des risques ISO 14971, permettant ainsi une évaluation continue des dangers, des modes de défaillance et de l'efficacité des mesures d'atténuation. Les données issues des réclamations, des événements indésirables, des rapports de tendances et du suivi clinique contribuent également à la gestion des CAPA et à l'amélioration continue.
04. Quel rôle jouent les données issues de la pratique clinique (RWE) dans la surveillance post-commercialisation ?
Les données issues de la pratique clinique (RWE) viennent renforcer le système de surveillance post-commercialisation (PMS) en fournissant des informations sur l'utilisation réelle des dispositifs, notamment les retours d'expérience des patients, les données de service, les registres et les sources de santé numériques. Les RWE permettent d'identifier les tendances, de valider les performances à long terme, d'étayer les mises à jour du rapport bénéfice/risque et d'éclairer les décisions cliniques ou réglementaires. Les autorités de réglementation attendent de plus en plus des fabricants qu'ils intègrent les RWE dans le PMS et dans l'évaluation continue post-commercialisation.
05. Dans quels cas une PMCF est-elle requise pour un dispositif médical ?
Une PMCF est requise lorsque les données cliniques existantes ne suffisent pas à confirmer la sécurité ou les performances à long terme, ou lorsque la technologie, les matériaux ou la destination d'utilisation d'un dispositif laissent entrevoir des risques potentiels à long terme. PMCF également déclenchée par de nouvelles tendances, des risques émergents, des incertitudes cliniques, de nouvelles conclusions issues de l'évaluation des risques pour la santé (HHE), ou lorsque les autorités de réglementation exigent la production continue de données pour les dispositifs à haut risque ou innovants.
06. Qu'est-ce qui déclenche un rappel de dispositif médical ou une mesure corrective sur le terrain (FCA) ?
Un rappel ou une action corrective et préventive (FCA) est déclenché lorsqu'un dispositif présente un risque potentiel ou avéré pour la sécurité, ne respecte pas les exigences réglementaires ou présente des problèmes de performance susceptibles de compromettre les résultats cliniques. Parmi les facteurs déclencheurs courants, on peut citer les tendances en matière de défauts, les incidents graves, les erreurs d'étiquetage, les défaillances de fabrication, les failles de cybersécurité ou de nouvelles données indiquant que le rapport bénéfice/risque du dispositif a changé.
07. Pourquoi Freyr est-il considéré comme un partenaire de premier plan pour les services de surveillance post-commercialisation ?
Freyr est largement reconnue pour son expertise approfondie en matière de réglementation, sa couverture mondiale des marchés et son approche structurée de la surveillance post-commercialisation (PMS), conforme aux exigences EU MDR, FDA et de la région Asie-Pacifique (APAC). Les organisations apprécient Freyr pour sa combinaison de connaissances sectorielles, de méthodologies fondées sur les données, de capacités multilingues et d'une expérience éprouvée dans la prise en charge de divers types de dispositifs, ce qui en fait un partenaire de confiance pour des opérations de surveillance post-commercialisation fiables et conformes.







