4 min de lecture
Le système de notification des dispositifs médicaux (MDR) est un outil de surveillance post-commercialisation utilisé par la Food and Drug Administration (FDA) pour contrôler les performances des dispositifs, détecter d'éventuels problèmes de sécurité liés à ces derniers et contribuer à l'évaluation du rapport bénéfice/risque des dispositifs. L'objectif du MDR est de détecter et de traiter rapidement les événements indésirables liés aux dispositifs. Il permet aux médecins, aux établissements de santé, aux fabricants et aux consommateurs de signaler volontairement ces événements afin de mieux comprendre la sécurité et l'efficacité des dispositifs après leur mise sur le marché.
Le MDR s'applique à toutes les classes de dispositifs médicaux, qu'ils soient fabriqués aux États-Unis d'Amérique (USA) ou importés aux USA. Les fabricants de dispositifs médicaux souhaitant commercialiser leurs dispositifs aux USA doivent se conformer au MDR, sous peine de sanctions financières. Il est applicable aux USA, y compris pour un événement étranger, c'est-à-dire qu'il s'applique aux dispositifs médicaux légalement commercialisés aux États-Unis, qu'ils soient fabriqués aux USA ou dans des pays étrangers. De plus, il existe divers cas d'applicabilité pour un MDR, tels que :
- si un dispositif est fabriqué aux US, distribué localement et sur d'autres marchés
- lorsqu'un dispositif est fabriqué aux US mais distribué sur d'autres marchés
- lorsqu'un dispositif est fabriqué à l'étranger, fourni aux US et sur d'autres marchés
- lorsqu'un dispositif est fabriqué à l'étranger et distribué localement, et
- lorsqu'un dispositif fait l'objet d'une enquête aux US
MDR et le flux du processus de déclaration
Le règlement MDR impose aux fabricants, aux importateurs et aux établissements utilisateurs de dispositifs de nombreuses obligations en matière de notification à la FDA de certains événements indésirables liés aux dispositifs et de problèmes liés aux produits. L'organigramme ci-dessous détaille étape par étape la procédure de notification.
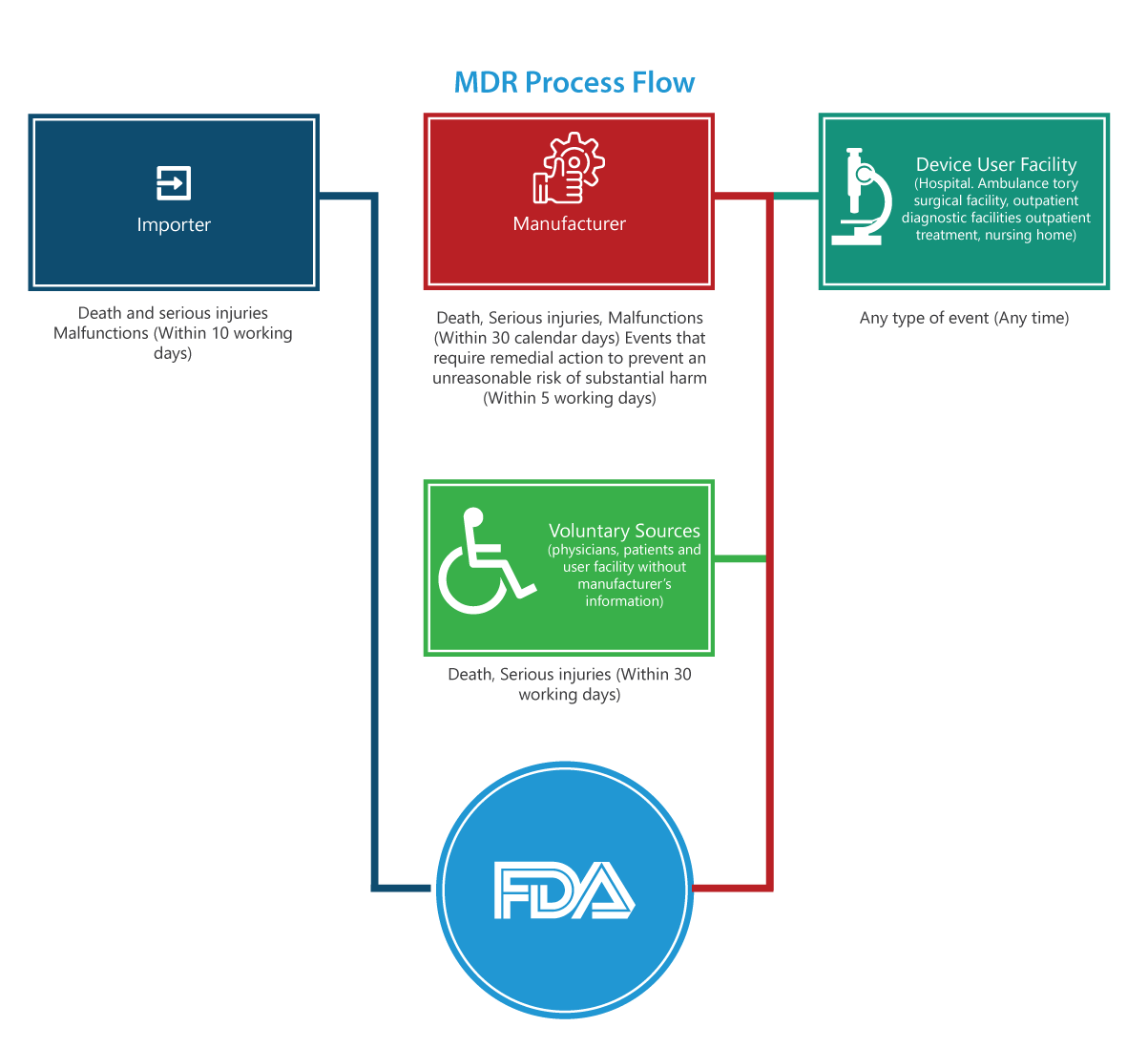
À qui cela s'applique-t-il ?
Importateurs
Les déclarations de décès, de blessures graves et de dysfonctionnements doivent être transmises à la FDA au fabricant dans un délai de 30 jours ouvrables. Si le dysfonctionnement est susceptible de causer des blessures ou des décès ailleurs, les importateurs sont tenus de le signaler au fabricant.
Fabricants.
Les déclarations relatives à un événement (décès, blessures graves et dysfonctionnements) désigné par FDA à un événement nécessitant des mesures correctives afin de prévenir un risque déraisonnable de préjudice grave pour la santé publique doivent être transmises à la FDA 5 jours ouvrables, en remplissant le formulaire 3500A.
Établissement utilisateur de dispositifs (Hôpital, établissement de chirurgie ambulatoire, maison de retraite, établissement de diagnostic ambulatoire ou établissement de traitement ambulatoire)
Les rapports doivent être transmis au fabricant du dispositif au plus tard dans les dix jours ouvrables suivant la date à laquelle l'établissement a pris connaissance d'informations indiquant qu'un dispositif a causé ou a pu causer, ou a contribué à causer, une blessure grave à un patient de l'établissement. Si le fabricant n'est pas connu, l'établissement doit transmettre le rapport à la FDA.
Groupes volontaires.
Les patients, les professionnels de santé et les consommateurs qui constatent un problème lié à un dispositif médical peuvent le signaler à FDA MedWatch
eMDR
En 2015, la FDA le système de notification électronique des incidents médicaux (eMDR) afin d'identifier les problèmes critiques liés à la qualité et à l'intégrité des données dans le cadre de la notification des incidents graves concernant toutes les catégories de dispositifs médicaux. L'eMDR est le mode de notification privilégié.
Les fabricants peuvent soumettre leur eMDR via une passerelle de soumission électronique (ESG). À compter de la soumission, la passerelle électronique peut mettre jusqu'à 48 heures pour envoyer un accusé de réception. En cas d'erreur lors de la soumission du rapport, un message s'affichera pour vous inviter à apporter les corrections nécessaires.
eMDR – En quoi est-il bénéfique ?
L'eMDR offre de multiples avantages par rapport au mécanisme de déclaration manuel (c'est-à-dire le MDR). Quelques avantages notables sur lesquels les fabricants / agences / patients peuvent compter sont énumérés ci-dessous :
- L'outil de soumission eMDR favorise une meilleure collaboration entre l'organisme, l'agence sanitaire (FDA) et les patients.
- L'eMDR permet de réduire les coûts. L'automatisation diminue les frais administratifs et les communications traditionnelles ; elle contribue à accélérer le processus et favorise un signalement efficace des événements, ce qui permet une interaction immédiate avec la FDA.
- Les processus manuels impliquent une paperasserie importante, peuvent être longs et difficiles à suivre et à traiter. La soumission eMDR est automatisée et centralisée. Les dossiers peuvent être récupérés facilement, ce qui permet de gagner beaucoup de temps lors de l'examen.
- L'eMDR permet aux parties concernées de signaler rapidement les erreurs de dépôt, ce qui évite les échanges manuels et fastidieux avec la FDA.
- eMDR sert de point d'entrée unique pour traiter toutes les soumissions électroniques dans un environnement hautement sécurisé ; son intérêt réside dans le fait que les plaintes adressées à l'organisation peuvent être directement associées au formulaire MedWatch et intégrées au portail FDA.
eMDR et le flux du processus de déclaration
Le règlement eMDR impose aux fabricants, aux importateurs et aux établissements utilisateurs de dispositifs médicaux l'obligation de signaler à la FDA certains événements indésirables liés aux dispositifs ainsi que certains problèmes liés aux produits. L'organigramme ci-dessous détaille étape par étape la procédure de notification.
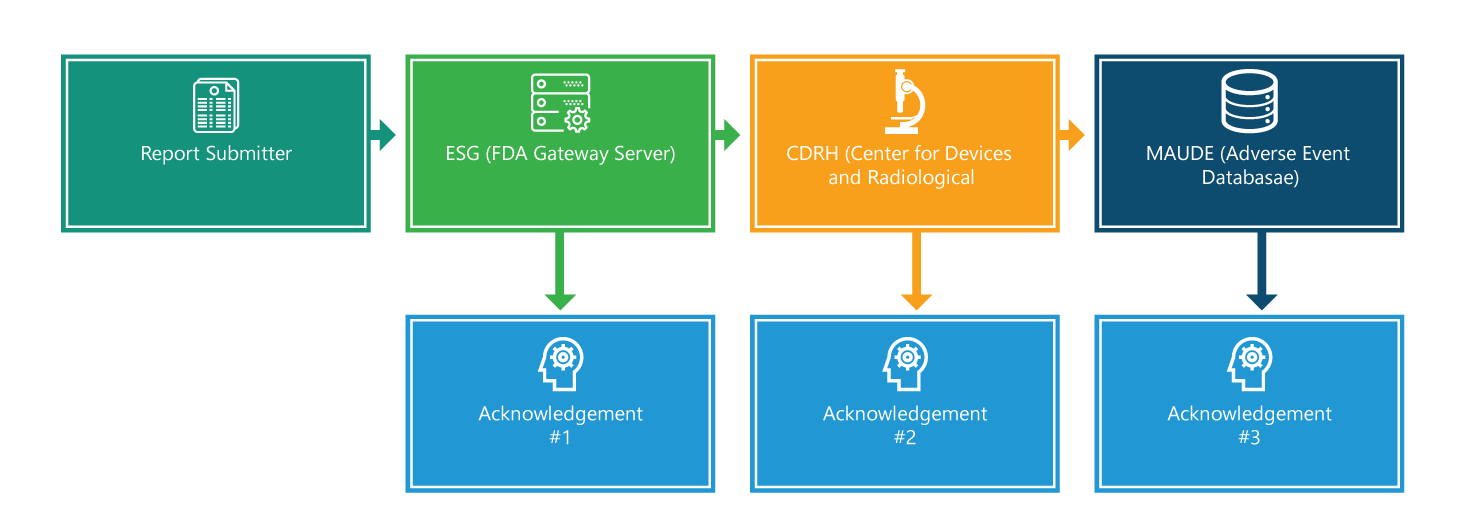
Le processus de déclaration comprend quatre étapes. À l'exception de la première étape, chaque étape est confirmée. De plus, chaque étape est accompagnée d'informations supplémentaires qui faciliteront le processus.
Étape 1 : Déclarant du rapport
Soumettre un eMDR. Au départ, pour effectuer une soumission, il faut disposer d'une signature électronique et s'assurer que les noms de fichiers de soumission ne contiennent qu'un seul point, utilisé pour indiquer l'extension du type de fichier (par exemple, 555.xml ou 555.pdf). Cependant, le délai de livraison et de traitement de la demande dépend de la taille globale de votre soumission ; les soumissions plus volumineuses prennent plus de temps à être livrées et traitées.
Étape 2 : Portail de soumission électronique (ESG)
Une fois votre déclaration transmise à ESG, vous devriez rapidement recevoir un accusé de réception n° 1, sauf si le système ESG indisponible pour cause de maintenance. Vous devez vérifier le statut de votre MDR sur le site ESG .
Étape 3 : CRDH
L'eMDR est automatiquement transmis par ESG Centre pour les dispositifs médicaux et la santé radiologique (CDRH). Une fois la transmission effectuée, comme à l'étape 2, vous devriez recevoir un accusé de réception, c'est-à-dire le n° 2.
Étape 4 : Expérience des dispositifs des fabricants et des utilisateurs (MAUDE)
Lorsque le CDRH valide et met à jour la soumission dans la base de données des événements indésirables (MAUDE), il est prévu que le déclarant reçoive un accusé de réception n°3. Il est à noter que toute erreur survenant lors de la validation et du chargement est enregistrée.
Le signalement des dispositifs médicaux (MDR) est un processus essentiel qui contribue à sauver des vies et à protéger les patients des risques inutiles. Il garantit que toutes les parties impliquées dans les soins aux patients sont responsables et vigilantes dans l'utilisation des dispositifs.
L'eMDR facilite la déclaration, mais la documentation et le suivi peuvent être gourmands en ressources. Faites-le correctement du premier coup ; parlez-nous à sales@freyrsolutions.com.