2 min de lecture
L'objectif principalFDAUS est d'examiner en permanence et de combler le fossé entre les processus réglementaires afin d'assurer l'importation et la commercialisation sans interruption de dispositifs médicaux nouveaux et de haute qualité sur le US .FDA 1998, laFDA US a publié un programme intitulé « Le nouveau paradigme 510(k) : approches alternatives pour démontrer l'équivalence substantielle dans les notifications préalables à la mise sur le marché ». Ce programme vise à établir une procédure efficace de soumission FDA (k) FDA , qui intègre certaines modifications par rapport à la demande 510(k) déjà approuvée. Cette nouvelle notification 510(k) propose trois types de soumissions, à savoir la 510(k) spéciale, la 510(k) abrégée et la 510(k) traditionnelle. En 2019, laFDA US a publié un document d'orientation sur la 510(k) spéciale décrivant une voie facultative pour les fabricants qui apportent certaines modifications bien définies à leur dispositif légalement commercialisé.
Pourquoi un 510(k) spécial ?
Lorsqu'un fabricant souhaite obtenir l'approbation pour les modifications qu'il a apportées à un dispositif déjà commercialisé, c'est-à-dire un dispositif existant, il peut demander un 510(k) spécial. Les principaux facteurs à prendre en compte pour déterminer si une modification apportée à un dispositif existant peut être éligible à un 510(k) spécial sont les suivants :
- Le changement concerne le propre dispositif prédicat légalement commercialisé du déclarant.
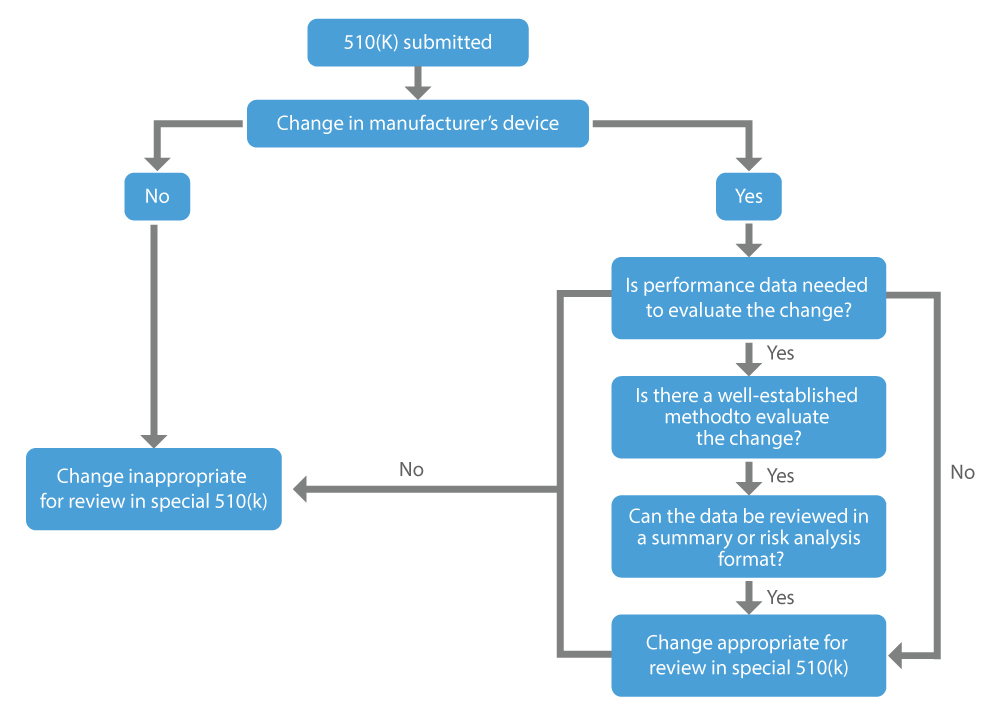
- Les données de performance ne sont pas requises, ou des méthodes bien établies sont disponibles si jugé nécessaire pour évaluer le changement.
- Toutes les données de performance pour étayer une détermination d'équivalence substantielle peuvent être examinées sous forme de résumé ou d'analyse des risques.
Documents requis pour le 510(k) spécial
- Lettre d'accompagnement
- Le nom du dispositif légalement commercialisé (existant) du fabricant et le numéro 510(k)
- Une description détaillée de la ou des modifications apportées au dispositif ayant entraîné la soumission d'un nouveau 510(k)
- Une comparaison du dispositif modifié au dispositif autorisé sous forme de tableau
- Autres modifications à l'étiquetage ou à la conception
- Un résumé concis des activités de contrôle de la conception.
- Sur la base de l'analyse des risques, une identification des activités de vérification et/ou de validation requises pour se conformer à la norme 21 CFR 820.30
- Formulaire d'indications d'utilisation
- Une déclaration attestant que le déclarant s'est conformé et n'est actuellement pas en violation des exigences de la procédure de contrôle de la conception, telles que spécifiées dans le 21 CFR 820.30, et que les dossiers sont disponibles pour examen sur demande.
Calendrier spécifique pour l'examen des demandes 510(k) par laFDA US
Conformément aux directives FDAintitulées « Politique de refus d'acceptation des demandes 510(k) », le délai d'examen des demandes 510(k) spéciales est de trente (30) jours à compter de leur réception.
Quand demander un 510(k) spécial ?
FDA US FDA des efforts constants pour garantir la mise à disposition de dispositifs médicaux sûrs et efficaces, dans le but de promouvoir la santé humaine. Le programme spécial 510(k) est efficace et conforme à la procédure d'examen la moins contraignante qui soit ; il aide les fabricants étrangers à commercialiser leurs dispositifs aux États-Unis et permet aux patients d'accéder rapidement aux nouveaux dispositifs médicaux.
Pour toute précision concernant la procédure spéciale 510(k) FDA, reach Freyr, un expert reconnu en matière de réglementation. Restez informé. Restez en conformité.