
3 minutes de lecture
Dans le secteur de la fabrication de dispositifs médicaux, en constante évolution et fortement réglementé, garantir la qualité des produits et la sécurité des patients n'est pas seulement une obligation légale, mais aussi un impératif stratégique. Des systèmes efficaces de gestion des réclamations et de vigilance, conformes à ISO 13485:2016, constituent la pierre angulaire de tout système de gestion de la qualité (SGQ) solide. Ces mécanismes garantissent la conformité aux réglementations internationales et renforcent la confiance des clients ainsi que la crédibilité de la marque.
Voyons comment la gestion des réclamations et la vigilance jouent un rôle fondamental dans le maintien de la qualité et de la conformité réglementaire dans le secteur des dispositifs médicaux, à la lumière des principes de ISO 13485:2016 et des cadres réglementaires internationaux.
Comprendre la gestion des réclamations selon ISO 13485:2016
ISO 13485:2016 définit une réclamation comme toute communication écrite, orale ou électronique faisant état de défauts liés à l'identité, à la qualité, à la durabilité, à la fiabilité, à la facilité d'utilisation, à la sécurité ou aux performances d'un dispositif médical. Cette norme impose aux fabricants de dispositifs médicaux de mettre en place des procédures documentées pour le traitement des réclamations, notamment l'évaluation, l'examen et la résolution de celles-ci, dans le cadre d'un système de gestion de la qualité efficace.
Un système de gestion des réclamations est essentiel pour garantir la conformité, maintenir la satisfaction des clients et améliorer la qualité des appareils. Ce processus doit être structuré et transparent, et comporter les étapes suivantes :
- Réception et documentation des réclamations
Toutes les réclamations doivent être enregistrées sans délai, en fournissant autant de détails que possible sur la nature du problème, les caractéristiques techniques et le modèle de l'appareil, les conditions d'utilisation, ainsi que les conséquences subies par l'utilisateur ou le patient. - Évaluation de l'obligation de déclaration
Une fois enregistrées, les plaintes doivent être évaluées afin de déterminer si elles constituent des incidents devant faire l'objet d'une déclaration. Aux États-Unis, par exemple, les fabricants de dispositifs médicaux sont tenus de signaler certains problèmes liés à leurs dispositifs dans le cadre du système de déclaration des dispositifs médicaux (MDR) réglementé par la FDA. Les fabricants doivent respecter les délais de déclaration prévus par la réglementation en vigueur. - Enquête et analyse des causes profondes
Toute plainte justifiée doit donner lieu à une enquête approfondie. Il est essentiel d'identifier la cause profonde pour mettre en œuvre des mesures correctives et préventives (CAPA) efficaces.
des mesures correctives et préventives (CAPA) Sur la base des conclusions relatives aux dispositifs médicaux, les entreprises doivent mettre en œuvre des mesures CAPA afin de résoudre le problème immédiat et d'éviter toute récidive, ainsi que de prévenir de futurs incidents.- Boucle de rétroaction et clôture
Une fois le problème résolu, la plainte doit être officiellement clôturée, accompagnée d'une documentation complète et d'un retour d'information au plaignant, si cela s'applique au dispositif médical.
La vigilance : un pilier essentiel de la surveillance post-commercialisation
La vigilance désigne le suivi et la notification des événements indésirables et des incidents survenant après la mise sur le marché d'un dispositif médical. Conformément à ISO 13485:2016 et aux réglementations internationales correspondantes, cette surveillance continue est essentielle pour détecter rapidement les risques potentiels et mettre en œuvre des interventions en temps opportun.
Le système de vigilance de l'Union européenne, tel que mis à jour par le règlement sur les dispositifs médicaux (EU MDR), impose aux fabricants de dispositifs médicaux de mettre en place des plans de surveillance post-commercialisation. Ces plans doivent inclure des procédures de collecte et d'analyse des données issues de l'utilisation réelle des dispositifs.
De même, en Inde, Central Drugs Standard Control Organization CDSCO) régit les procédures de vigilance et de traitement des plaintes concernant les dispositifs médicaux. Elle encourage la notification des événements indésirables et des défaillances de produits afin d'améliorer en permanence la sécurité et l'efficacité des dispositifs médicaux.
Les éléments clés d'un système de vigilance efficace sont les suivants :
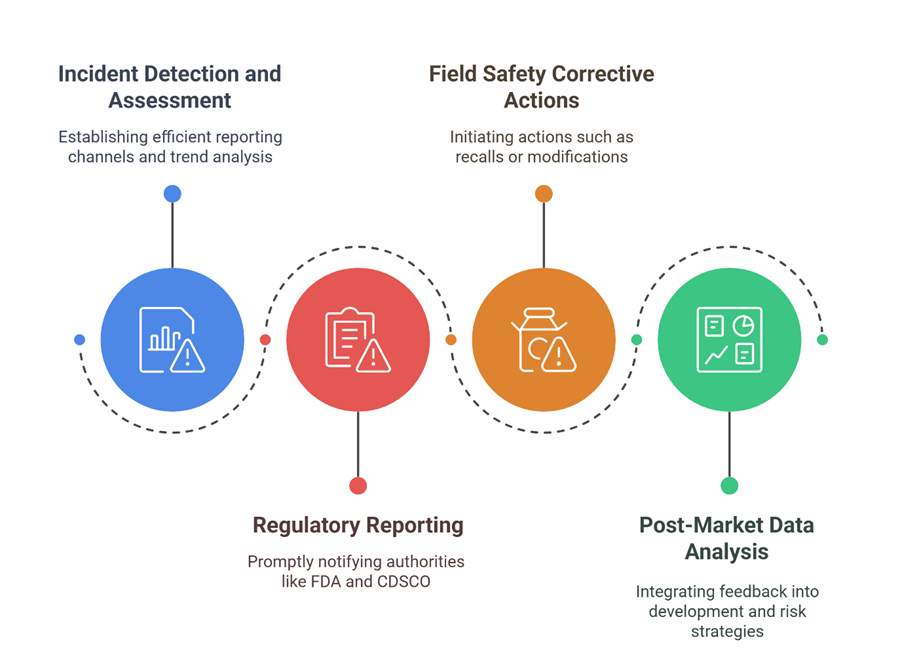
Élaboration d'une stratégie intégrée de gestion des plaintes et de vigilance
Les fabricants de dispositifs médicaux peuvent améliorer leur système de gestion de la qualité (SGQ) en intégrant la gestion des réclamations et la vigilance au sein d'un système unique et cohérent. Voici quelques bonnes pratiques :
Des procédures opérationnelles standard (SOP) et des descriptions de poste claires
Les procédures opérationnelles standard (SOP) doivent décrire les étapes à suivre pour la réception, l'évaluation, le signalement et le règlement des réclamations. Elles précisent qui est responsable à chaque étape afin d'éviter les retards et les lacunes.
- Formation et sensibilisation du personnel
Une formation régulière du personnel permet de s'assurer que les employés savent identifier et gérer efficacement les plaintes et les événements indésirables. - Outils numériques et automatisation
L'utilisation d'un logiciel de gestion de la qualité (QMS) permet de rationaliser le suivi des réclamations, la documentation et le reporting. L'automatisation améliore la traçabilité et la conformité à ISO 13485:2016. - Culture de la qualité et amélioration continue
La promotion d'une philosophie qui valorise le retour d'information encourage le signalement proactif des problèmes et favorise l'innovation en matière d'amélioration de la qualité.
Au-delà de la conformité : un atout marketing
La mise en place de systèmes conformes à ISO 13485:2016 pour la gestion des réclamations et la vigilance permet non seulement de garantir le respect de la réglementation, mais aussi de positionner l'entreprise comme une entité digne de confiance et soucieuse de la qualité. Dans un secteur où la sécurité et la performance ont un impact direct sur la vie des personnes, un tel engagement peut constituer un puissant outil marketing.
Selon FDA la mise en conformité avec les normes internationales facilite l'accès aux marchés mondiaux, renforce la confiance des clients et réduit le risque de rappels de produits ou de litiges coûteux.
| Région | Cadre réglementaire | Référence n° / Article relatif au traitement des réclamations | Référence n° / Article relatif à la vigilance / Déclaration des événements indésirables |
| Union européenne | EU MDR 2017/745 | Article 83, annexe III (système PMS) ; article 10, paragraphe 9 | Articles 87 à 92 (Système de vigilance), annexe III |
| États-Unis (FDA) | 21 CFR, partie 820 (QSR) | §820.198 – Dossiers de réclamations | 21 CFR Partie 803 – Déclaration des dispositifs médicaux (MDR) |
| Inde (CDSCO) | Règlement sur les dispositifs médicaux de 2017 (MDR) | Chapitre VII, articles 25 et 26 | Chapitre VII, article 27 |
| International (norme ISO) | ISO 13485:2016 | Article 8.2.2 – Traitement des réclamations | Clause 8.2.3 – Déclaration aux autorités de réglementation |
Conclusion
Dans le contexte réglementaire actuel, très concurrentiel, du secteur des dispositifs médicaux, la mise en place de systèmes efficaces de gestion des réclamations et de vigilance est indispensable. Conformes à ISO 13485:2016, ces processus permettent non seulement de protéger les patients, mais aussi de bâtir une marque solide, fondée sur la réputation. En investissant dans des procédures systématiques, une formation continue et une vigilance post-commercialisation, les fabricants peuvent transformer les exigences réglementaires en opportunités de croissance et de leadership sur le marché.









