
2 min de lecture
FDA US FDA publié un document d'orientation destiné à aider les acteurs du secteur et le personnel de l'Agence de santé (HA) à déterminer dans quels cas une modification logicielle apportée à un dispositif médical oblige le fabricant à soumettre et à obtenir FDA par le biais d'une nouvelle notification préalable à la mise sur le marché (510(k)). Ces lignes directrices visent à améliorer la prévisibilité, la cohérence et la transparence du processus décisionnel relatif au moment de la soumission, en proposant une approche la moins contraignante possible et en décrivant le cadre réglementaire, les politiques et les pratiques qui sous-tendent une telle décision, notamment en ce qui concerne les modifications logicielles. Découvrons en détail ces FDA .
Principes FDA et organigramme FDA
Afin d'aider les fabricants de dispositifs médicaux à appliquer les principes fondamentaux, ce document fournit un organigramme, des précisions supplémentaires et des exemples nécessaires à la prise de décision concernant une nouvelle notification préalable à la mise sur le marché (510(k)) pour une modification logicielle apportée à un dispositif déjà approuvé aux US. De plus, il convient de suivre plusieurs principes directeurs lors de l'utilisation de ce guide pour déterminer s'il faut soumettre une nouvelle notification 510(k) afin de modifier un dispositif existant. Certains d'entre eux sont largement connus et découlent de la politique actuelle FDA (k), tandis que d'autres sont nécessaires pour appliquer le schéma logique mentionné dans ce guide. Conformément au guide, le schéma fourni ne peut pas couvrir toutes les subtilités possibles liées à de telles modifications et leur incidence sur la décision. Par conséquent, pour déterminer la nécessité d'une nouvelle notification préalable à la mise sur le marché 510(k), les fabricants de dispositifs médicaux doivent tenir compte des principes généraux et de l'organigramme résumés ci-dessous.
- Modifications qui affectent la sécurité ou l'efficacité d'un dispositif
- Évaluation initiale basée sur les risques
- Conséquences imprévues des changements
- Gestion des risques
- Rôle des tests (activités de vérification et de validation) pour évaluer si un changement pourrait affecter de manière significative la sécurité et l'efficacité
- Évaluer les changements simultanés pour déterminer si la soumission d'un nouveau 510(k) est requise
- Dispositif comparatif approprié et l'effet cumulatif des changements
- Exigence documentaire (21 CFR Part 820)
- Soumissions 510(k) pour les dispositifs modifiés
- Déterminations d'équivalence substantielle
- La soumission d'un nouveau 510(k) est probablement requise si un fabricant modifie son dispositif de manière à affecter sa sécurité ou son efficacité. Cependant, les modifications qui ne sont pas censées affecter la sécurité ou l'efficacité d'un dispositif doivent tout de même être évaluées.
- Pour déterminer si un changement ou une modification pourrait affecter de manière significative la sécurité ou l'efficacité, le fabricant doit d'abord effectuer une évaluation basée sur les risques afin de savoir si le changement pourrait affecter la sécurité ou l'efficacité du dispositif, positivement ou négativement. Cette évaluation basée sur les risques doit identifier et analyser tous les nouveaux risques et les changements dans les risques existants résultant de la modification du dispositif et conduire à une décision initiale quant à la nécessité de soumettre un nouveau 510(k).
- Parfois, des conséquences supplémentaires imprévues ou non planifiées peuvent être déclenchées lors des soumissions de logiciels. L'organigramme doit évaluer ces conséquences pour déterminer si la soumission d'un nouveau 510(k) est nécessaire.
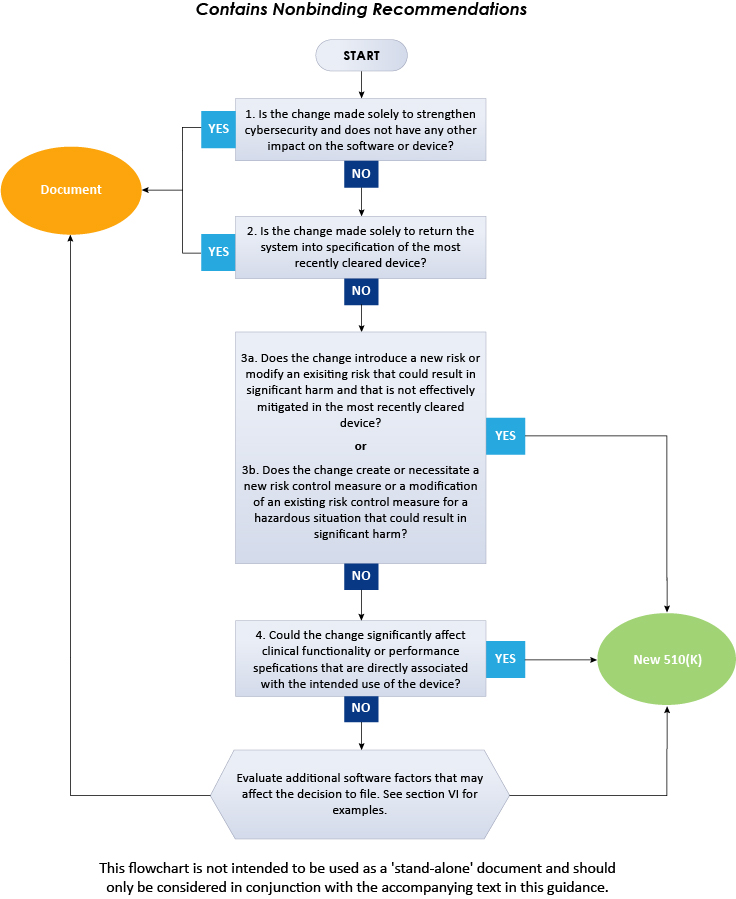
L'organigramme ci-dessus présente la procédure étape par étape à suivre pour déterminer s'il convient de déposer une demande 510(k) pour des modifications logicielles apportées à des dispositifs existants. En conclusion, les présentes FDA décrivent en détail l'approche que doivent suivre les fabricants de dispositifs médicaux lorsqu'ils doivent déterminer si les modifications logicielles apportées à un dispositif médical existant nécessitent le dépôt d'une nouvelle demande 510(k). Pour en savoir plus sur les FDA , consultez Freyr, un expert reconnu en matière de réglementation. Restez informé. Restez en conformité.









