
Was ist QMSR?
Der FDA ist ein gestraffter Ansatz, der den Anforderungen ISO 13485:2016 entspricht und eine Aktualisierung der früheren QSR-Struktur darstellt. Diese Angleichung ist von entscheidender Bedeutung, da sie die weltweite Einhaltung der Vorschriften für Hersteller vereinfacht, insbesondere für diejenigen, die global tätig sind. Diese Harmonisierung ermöglicht es Unternehmen, die regulatorischen Anforderungen sowohl in den US in anderen Märkten auf wesentlich einheitlichere Weise zu erfüllen.
Die QMSR schreibt Verbesserungen im Risikomanagement, bei der Produktkonstruktion und bei der Überwachung nach dem Inverkehrbringen vor. Diese Aktualisierung mag zwar zu einer gewissen Komplexität führen, bietet den Herstellern jedoch auch die Möglichkeit, ihre Qualitätsverfahren zu standardisieren, was wiederum die Produktsicherheit erhöht und zu einer besseren Dokumentation führt, was bei USFDA von entscheidender Bedeutung sein kann.
Warum ist der Übergang von QSR zu QMSR so entscheidend?
![]()
![]()
Anpassung an globale Standards
Dies wird den Herstellern dabei helfen, sich durch die Umstellung auf QMSR an die globalen Anforderungen an Qualitätsmanagementsysteme für Medizinprodukte anzupassen, wodurch regulatorische Doppelarbeit für weltweit tätige Hersteller von Medizinprodukten reduziert wird.
![]()
Verbesserte Qualität, Sicherheit und Wirksamkeit
Diese Integration gewährleistet, dass die Produkte gemäß den weltweiten Anforderungen an das Qualitätsmanagementsystem hergestellt werden. Dies hilft dem Hersteller, die Qualität und Sicherheit der Produkte zu verbessern.
![]()
Regulatorische Compliance
Dies hilft den Herstellern, sich an die neuen Anforderungen anzupassen, und verringert das Risiko von Verstößen, wenn die Verordnung am 2. Februar 2026 in Kraft tritt.



Wichtige Änderungen beim Übergang von QSR zu QMSR:
![]()
Angleichung an die Terminologie der ISO 13485:2016
Dadurch wird sichergestellt, dass Hersteller weltweit anerkannte Standards anwenden.
![]()
Risikomanagement:
Der Schwerpunkt liegt auf dem Risikomanagement während des gesamten Lebenszyklus des Medizinprodukts.
![]()
Geräteaufbau und Bedienelemente
Im Rahmen FDA wurden die Konstruktionskontrollen ausgeweitet, um sicherzustellen, dass die Hersteller den Bedürfnissen der Anwender, der Sicherheit des Produkts und den Leistungskriterien in vollem Umfang Rechnung tragen; dies ist ein Schwerpunkt bei USFDA
![]()
Post-Market Surveillance
Unternehmen müssen das System zur Überwachung nach dem Inverkehrbringen verbessern. Dies bedeutet, dass Hersteller Informationen über die Sicherheit und Wirksamkeit der Produkte sammeln müssen, um Probleme rasch zu erkennen und zu beheben.
![]()
Dokumentation und Aufbewahrung von Unterlagen
In der endgültigen Regelung wird betont, dass eine lückenlose Dokumentation von entscheidender Bedeutung ist, insbesondere während der Inspektion.
Wichtige Termine
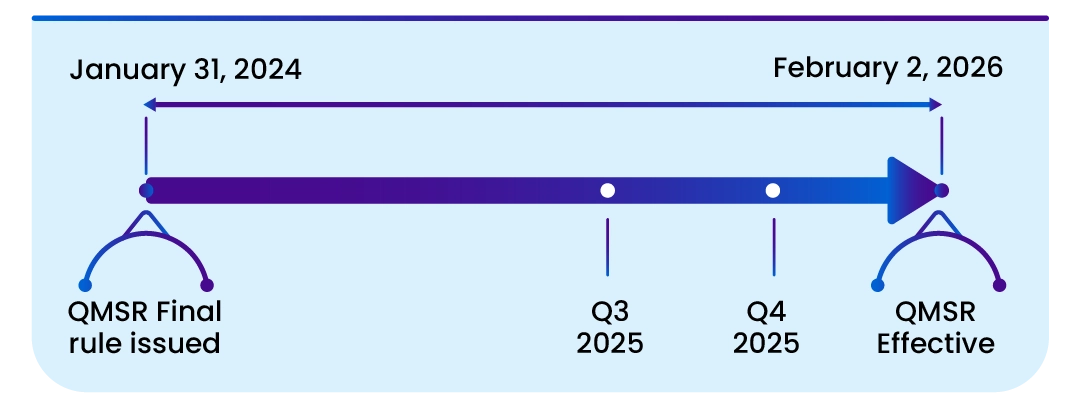
Diese Vorschrift tritt am 2. Februar 2026 in Kraft. Die Einbeziehung bestimmter in dieser Vorschrift aufgeführter Unterlagen durch Verweis wurde vom Direktor des Federal Register am 2. Februar 2024 genehmigt.
Holen Sie sich fachkundige Beratung zur Einhaltung der QMSR-Vorschriften
Warum sollten Sie sich an das FDA -Rahmenwerk FDA halten?
![Regulatorische Intelligenz (RI)]() Die QMSR tritt am 2. Februar 2026 in Kraft; Hersteller sind verpflichtet, ihr Qualitätsmanagementsystem entsprechend anzupassen.
Die QMSR tritt am 2. Februar 2026 in Kraft; Hersteller sind verpflichtet, ihr Qualitätsmanagementsystem entsprechend anzupassen.![Ein einziger Partner für alles]() Eine fristgerechte Umsetzung und eine effektive Wartung können das Risiko von Produktrückrufen und Beschwerden verringern.
Eine fristgerechte Umsetzung und eine effektive Wartung können das Risiko von Produktrückrufen und Beschwerden verringern.![Gewährleistung der Einreichungsgenauigkeit]() Die Einhaltung der QMSR-Vorschriften kann dazu führen, dassFDA US FDA weniger Beanstandungen vorbringt,FDA sich nicht auf den Status des Produkts auf dem regulierten Markt auswirkt.
Die Einhaltung der QMSR-Vorschriften kann dazu führen, dassFDA US FDA weniger Beanstandungen vorbringt,FDA sich nicht auf den Status des Produkts auf dem regulierten Markt auswirkt.
 Die QMSR tritt am 2. Februar 2026 in Kraft; Hersteller sind verpflichtet, ihr Qualitätsmanagementsystem entsprechend anzupassen.
Die QMSR tritt am 2. Februar 2026 in Kraft; Hersteller sind verpflichtet, ihr Qualitätsmanagementsystem entsprechend anzupassen. Eine fristgerechte Umsetzung und eine effektive Wartung können das Risiko von Produktrückrufen und Beschwerden verringern.
Eine fristgerechte Umsetzung und eine effektive Wartung können das Risiko von Produktrückrufen und Beschwerden verringern. Die Einhaltung der QMSR-Vorschriften kann dazu führen, dassFDA US FDA weniger Beanstandungen vorbringt,FDA sich nicht auf den Status des Produkts auf dem regulierten Markt auswirkt.
Die Einhaltung der QMSR-Vorschriften kann dazu führen, dassFDA US FDA weniger Beanstandungen vorbringt,FDA sich nicht auf den Status des Produkts auf dem regulierten Markt auswirkt.Entscheiden Sie sich für die FDA Beratungsleistungen von Freyr, bei denen unsere erstklassigen Berater Sie bei jedem Schritt des Lebenszyklus Ihres Medizinprodukts sorgfältig begleiten, um eine reibungslose Umsetzung der QMSR-Anforderungen zu gewährleisten!
Holen Sie sich fachkundige Beratung zur Einhaltung der QMSR-Vorschriften
QMSR
- Klassifizierung von Medizinprodukten gemäß derFDA.
- Erstellung von Dokumenten gemäß 21 CFR 820.
- Lückenanalyse der bestehenden Dokumente des Qualitätsmanagementsystems (QMS) gemäß 21 CFR 820.
- Korrekturplan für die Einhaltung von 21 CFR 820.
- Probeprüfungen.
- Ein engagiertes Qualitätssicherungsteam (QA) von Experten für Medizinprodukte.
- Nachgewiesene Expertise im Umgang mit der Einhaltung von 21 CFR 820.
- Flexible Modelle für die Projektabwicklung.