5 min de lecture
Soumissions prévues pour le 24 sept. 2016.
Maintenant que la deuxième phase de mise en conformité avec l'UDI, concernant les dispositifs médicaux de classe III I/LS/LS, a été mise en œuvre, de nombreux fabricants de dispositifs, en particulier ceux de classe II, se demandent comment se préparer au mieux à la date limite de soumission des données pour les dispositifs de classe II, fixée au 24 septembre 2016. Afin de les aider à s'y préparer, chez Freyr, nous avons identifié certaines des conditions préalables qu'ils doivent prendre en compte pour mettre leurs dispositifs de classe II en conformité avec l'obligation UDI FDA.
La nouvelle réglementation impose que tous les dispositifs médicaux de classe II soient étiquetés et conditionnés avec un identifiant unique de dispositif (UDI) et enregistrés dans la base de données mondiale d'identification unique des dispositifs (GUDID) FDA. Compte tenu de l'évolution constante des exigences de conformité et des délais de soumission de plus en plus courts, le défi pour les fabricants de dispositifs consiste à maîtriser les subtilités des processus de conformité. Parallèlement, ils doivent s'assurer qu'aucune des caractéristiques clés des dispositifs n'est omise lorsqu'ils rassemblent les données dispersées dans différents systèmes et les harmonisent dans des feuilles de calcul pour créer des rapports de conformité.
Afin d'aider les fabricants de dispositifs à naviguer facilement et sans erreurs à travers ce processus de conformité complexe et critique en termes de temps, Freyr a compilé les prérequis suivants à respecter.
Déterminer la date de mise en conformité avec l'UDI: depuis la FDA la réglementation définitive FDA , certaines dates de mise en conformité des dispositifs ont été modifiées et reportées. Afin de planifier minutieusement les stratégies et les processus de mise en conformité et d'éviter des modifications précipitées de dernière minute, les étiqueteurs doivent déterminer la date exacte de mise en conformité.
Date de conformité des dispositifs de classe II Exigences en matière de conformité 24 sept. 2016Les dispositifs de classe III devant être étiquetés avec un UDI doivent porter un UDI comme marquage permanent sur le dispositif lui-même si le dispositif est destiné à être utilisé plus d'une fois et à être retraité avant chaque utilisation. Les étiquettes et les emballages des dispositifs médicaux de classe II doivent porter un UDI. Les dates figurant sur les étiquettes de ces dispositifs doivent être formatées comme requis Les logiciels autonomes de Classe II doivent fournir leur UDI conformément aux exigences. Les données pour les dispositifs de classe II qui doivent être étiquetés avec un UDI doivent être soumises à la base de données GUDID Pour la plupart des dispositifs, la date de conformité pour le marquage direct est différente des autres exigences. En fonction de la catégorie de votre produit, qu'il soit destiné à être réutilisé ou retraité, déterminez la date de conformité UDI pour le marquage direct comme indiqué ici :
Dates de conformité pour le marquage direct Catégorie de dispositif – Réutilisé et Retraité 24 sept. 2015 Dispositifs de maintien et de soutien des fonctions vitales, quelle que soit leur classe 24 sept. 2016 Dispositifs de classe III et dispositifs autorisés en vertu de la loi sur les services de santé publique 24 sept. 2018 Dispositifs de classe II 24 sept. 2020 Dispositifs de classe I et dispositifs non classés Évaluer la nécessité du marquage direct du numéro UDI : Tous les dispositifs médicaux utilisés plus d'une fois ou devant être retraités avant chaque utilisation doivent porter un marquage direct de l'UDI. L'exception concerne les dispositifs implantables qui ne nécessitent pas de marquage direct selon la règle UDI. Les dispositifs à usage unique, même s'ils sont retraités, ne sont pas non plus tenus de porter un UDI permanent – 21 CFR 801.45(d)(3). Ainsi, évaluez la nécessité du marquage direct en fonction de la catégorie de dispositifs médicaux que vous fabriquez.
- Plan de conformité complète: Passez en revue les FDA auxquelles vos produits spécifiques doivent se conformer. Réalisez une analyse approfondie des lacunes afin d'identifier les insuffisances en matière de données ou de technologie, ce qui vous permettra de relever certains des principaux défis liés au respect des FDA stricts FDA . Parmi ces défis, on peut citer l'obtention des informations DI ou PI, ainsi que le traitement de grands volumes de données non structurées provenant de sources disparates, etc. Au lieu de travailler d'arrache-pied jusqu'à tard dans la nuit pour harmoniser toutes les données relatives aux dispositifs médicaux à la dernière minute, planifiez à l'avance une conformité complète grâce à des systèmes et des outils validés qui prennent en charge l'intégration, la qualité et la gestion des données.
![]()
Obtenir un numéro DI et l'adhésion à l'Agence : L'UDI se compose d'un identifiant de dispositif (DI – numéro unique basé sur la version ou le modèle du dispositif) et d'un identifiant de produit (PI – comprenant le numéro de lot, le numéro de série ou la date d'expiration). La partie DI de l'UDI servira de clé primaire pour rechercher des informations sur le dispositif dans le GUDID. Pour attribuer le DI, la FDA accrédité trois agences émettrices : GS1, HIBCC et ICCBBA. Dans ce scénario, les étiqueteurs doivent adhérer à l'une de ces agences pour obtenir le numéro DI qui doit être saisi dans le GUDID FDA.
![]()
Identifiant Attributs Organismes émetteurs UDI DI (Identifiant de dispositif – Données statiques)
Doit être synchronisé avec GUDIDNombre unique de
Fabricant
Marque du dispositif
Modèle du dispositif
GSI
HIBCC
ICCBBAPI (Identifiant de Produit – Données Dynamiques)
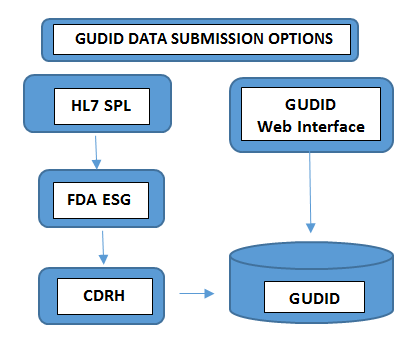
Obligatoire sur tous les niveaux d'emballageNuméro de lot, Numéro de série, Date de fabrication, Date de péremption, - Soumettre les données: La manière dont vous transmettez les données au GUDID dépend du volume de votre portefeuille de produits. Les fabricants de dispositifs disposant d’un nombre réduit de dispositifs choisissent de transmettre les informations UDI manuellement via l’interface Web gratuite du GUDID FDA. Dans ce cas, un seul enregistrement DI peut être transmis à la fois via une interface Web sécurisée du GUDID.Dans l'autre cas, les fabricants disposant d'un plus grand nombre de gammes de produits choisissent l'option de soumission HL7 SPL pour collecter les données par voie électronique et convertir les données consolidées au format SPL avant de les soumettre à la passerelle de soumission électronique (ESG) FDA, en utilisant le numéro DUNS. Veuillez noter que le compte GUDID n'est pas lié au type de soumission. Le compte sert à vous identifier, vous, l'étiqueteur, afin de permettre la soumission des données relatives aux dispositifs via les deux options.
![]()
- Créer un compte GUDID: Un étiqueteur ou un fabricant de dispositifs médicaux doit disposer d'un ou plusieurs comptes GUDID en fonction du nombre de rôles à attribuer ; par exemple, coordinateur GUDID, utilisateur chargé de la saisie des données, etc. Cependant, pour autoriser chaque rôle à la saisie des données, le fabricant doit obtenir l'accord de la FDA la création du compte. La procédure de création d'un compte GUDID approprié consiste à envoyer une demande par e-mail à FDA quoi le demandeur, c'est-à-dire vous, recevra un formulaire de demande de compte à remplir. Une fois que vous aurez renvoyé le formulaire rempli à la FDA e-mail, l'agence examinera le formulaire, puis vous enverra un e-mail contenant les informations de connexion au compte GUDID.


La mise en œuvre de l'UDI est un processus complexe et chronophage. Au cours de ce processus, tout en se conformant aux exigences FDAl'UDI FDA, les fabricants de dispositifs médicaux sont confrontés à de nombreux défis liés à la gestion, à l'intégration et à la transmission des données. La date limite de mise en conformité pour les dispositifs de classe II n'étant plus qu'à un an, Freyr recommande aux entreprises de commencer dès maintenant à s'y préparer.
Pour guider votre organisation à travers ce processus de conformité complexe, Freyr offre le meilleur des deux mondes – une solution logicielle UDI à la demande, entièrement configurable, Freyr IDENTITY, ainsi qu'un Centre d'Excellence (CoE) qui propose des services UDI de premier ordre, rentables et personnalisables, conçus pour répondre à vos exigences uniques et spécifiques.









