2 min de lecture
Un « dispositif de référence » est un dispositif médical qui a déjà été approuvé par la US and Drug Administration (FDAUS et qui est déjà commercialisé ; il sert de référence pour les nouveaux dispositifs médicaux dont l'autorisation est demandée dans le cadre de la procédure d'autorisation510(k) FDA.
Le dispositif en question doit être prouvé au moins aussi sûr et efficace que le dispositif de référence en termes d'utilisation prévue et de caractéristiques technologiques. Cette comparaison est connue sous le nom de détermination de l'« équivalence substantielle ».
Un nouveau dispositif n'a pas besoin d'être identique au dispositif de référence pour être considéré comme substantiellement équivalent à celui-ci.
Comment identifier un dispositif de référence ?
La base de données FDAfournit un code produit à trois lettres pour chaque classification de dispositif. La base de données FDA (k) contient des informations sur tous les dispositifs autorisés via laprocédure 510(k). Une fois que vous disposez du code produit à trois lettres, vous pouvez obtenir une liste de tous les produits, de toutes les entreprises et des noms commerciaux de tous les concurrents ou concurrents potentiels que vous souhaitez examiner. Vous pouvez ensuite effectuer une analyse et une comparaison approfondies afin de sélectionner un dispositif de référence.
Vous trouverez ci-dessous un organigramme illustrant le processus d'identification et de sélection d'un dispositif de référence.
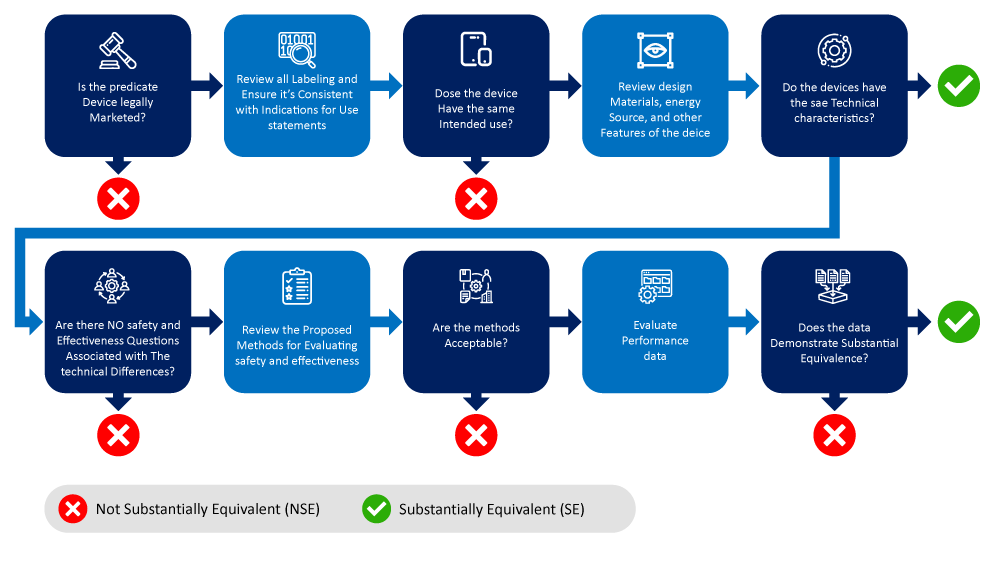
Facteurs à prendre en compte lors de la détermination du ou des dispositifs de référence
- Utilisation prévue : L'utilisation prévue après le dispositif de référence doit être similaire à celle du nouveau dispositif. Par exemple, si le nouveau dispositif est destiné à la surveillance cardiaque, le dispositif de référence doit également être un dispositif de surveillance cardiaque.
- Caractéristiques technologiques : Le dispositif de référence doit être identique au nouveau dispositif en termes de caractéristiques technologiques. Prenez, par exemple, la conception, les matériaux utilisés et le mode de fonctionnement qui devraient être similaires.
- Biocompatibilité : Les évaluations de biocompatibilité d'un dispositif médical ou d'un composant ne doivent pas se limiter aux matières premières utilisées dans le dispositif et le processus de fabrication, et des produits chimiques supplémentaires doivent également être pris en compte. Ce facteur, cependant, ne s'applique pas aux DIV.
- Dernière technologie : Le dispositif de référence ne doit pas être obsolète et doit représenter la dernière technologie médicale.
Le dispositif de référence est un facteur clé pour déterminer si un nouveau dispositif médical peut être mis sur le marché via la procédure 510(k). Choisir le mauvais dispositif de référence pourrait entraîner un processus d'approbation réglementaire plus coûteux et plus long, tandis que choisir le bon dispositif de référence peut aider à réduire les coûts et le temps nécessaires pour mettre un nouveau dispositif médical sur le marché. Si le dispositif de référence n'est pas adapté, cela pourrait entraîner des retards et des dépenses supplémentaires.
Pour obtenir de l'aide concernant le processus de soumission 510(k) de votre dispositif médical, planifiez un appel avec les experts en réglementation de Freyr, qui peuvent vous aider à naviguer dans les procédures. Restez informé. Restez conforme.









