2 Min. Lesezeit
Das zentrale Anliegen der US FDA ist es, die regulatorischen Prozesse für den ununterbrochenen Import und Verkauf neuer und hochwertiger Medizinprodukte auf dem US-Markt ständig zu prüfen und bestehende Lücken zu schließen. Die US FDA veröffentlichte 1998 ein Programm mit dem Titel „Das neue 510(k)-Paradigma: Alternative Ansätze zum Nachweis der wesentlichen Äquivalenz in Prämarkt-Benachrichtigungen“. Es zielt darauf ab, einen effizienten FDA 510(k)-Einreichungsweg zu etablieren, der bestimmte Änderungen in der bereits genehmigten 510(k)-Anwendung berücksichtigt. Diese neue 510(k)-Benachrichtigung bietet drei Arten von Einreichungen an: spezielle 510(k), abgekürzte 510(k) und traditionelle 510(k). Im Jahr 2019 veröffentlichte die US FDA ein spezielles 510(k)-Leitliniendokument, das einen optionalen Weg für Hersteller beschreibt, die bestimmte klar definierte Änderungen an ihrem rechtmäßig vermarkteten Produkt vornehmen.
Warum ein Special 510(k)?
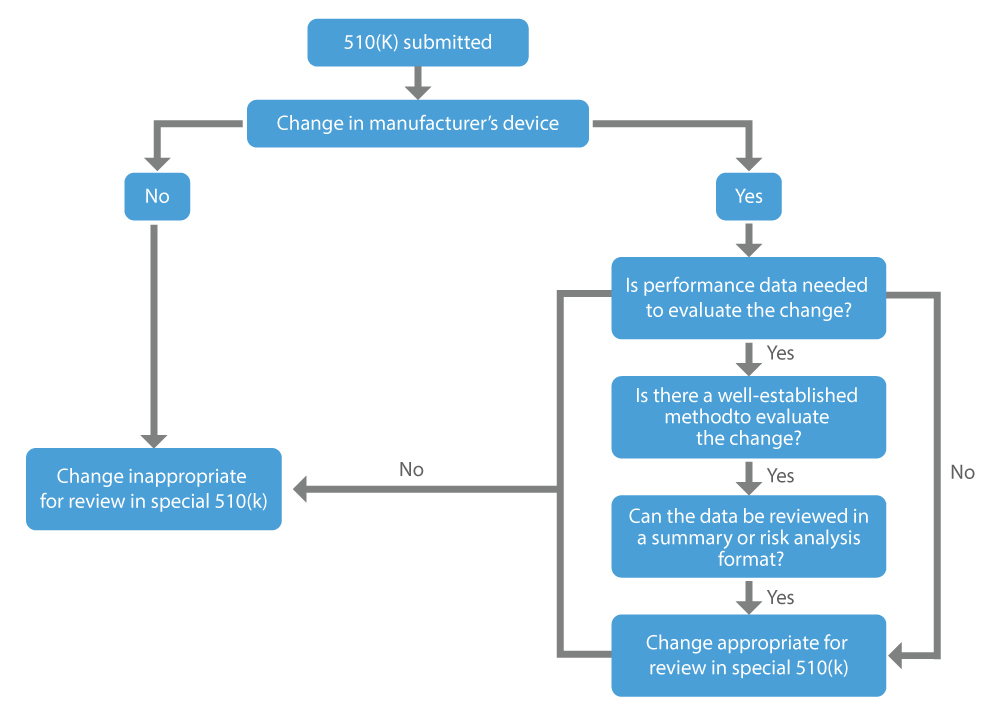
Wenn ein Hersteller die Genehmigung für Änderungen an einem bereits auf dem Markt befindlichen Gerät, d.h. dem bestehenden Gerät, beantragen möchte, kann er einen Special 510(k) beantragen. Die wichtigsten Faktoren, die bei der Entscheidung, ob eine Änderung an einem bestehenden Gerät für einen Special 510(k) geeignet ist, zu berücksichtigen sind, sind die folgenden:
- Die Änderung betrifft das eigene, rechtmäßig vermarktete Referenzgerät des Antragstellers.
- Leistungsdaten sind nicht erforderlich, oder es stehen etablierte Methoden zur Verfügung, falls dies zur Bewertung der Änderung als notwendig erachtet wird.
- Alle Leistungsdaten zur Unterstützung einer Bestimmung der wesentlichen Gleichwertigkeit können in einem zusammenfassenden Format oder als Risikoanalyse überprüft werden.
Erforderliche Dokumente für Special 510(k)
- Anschreiben
- Der Name des rechtmäßig vermarkteten (bestehenden) Produkts des Herstellers und die 510(k)-Nummer
- Eine detaillierte Beschreibung der Änderung(en) am Gerät, die zur Einreichung einer neuen 510(k) führte(n)
- Ein Vergleich des modifizierten Geräts mit dem zugelassenen Gerät in Tabellenform
- Weitere Änderungen an Kennzeichnung oder Design
- Eine prägnante Zusammenfassung der Designkontrollaktivitäten
- Basierend auf der Risikoanalyse, eine Identifizierung der Verifizierungs und/oder Validierungsaktivitäten, die zur Einhaltung von 21 CFR 820.30 erforderlich sind.
- Formular für Anwendungsgebiete
- Eine Erklärung, dass der Einreicher die Anforderungen erfüllt hat und derzeit nicht gegen die Anforderungen des Designkontrollverfahrens verstößt, wie in 21 CFR 820.30 festgelegt, und dass die Aufzeichnungen auf Anfrage zur Überprüfung verfügbar sind.
Zeitplan für die spezielle 510(k)-Prüfung durch die US FDA
Gemäß den Richtlinien der FDA „Refuse to Accept Policy for 510(k)s“ beträgt die Überprüfungsfrist für spezielle 510(k)-Einreichungen dreißig (30) Tage nach deren Eingang.
Wann ist eine spezielle 510(k) zu beantragen?
Die US FDA unternimmt konsequente Anstrengungen, um sichere und wirksame Medizinprodukte bereitzustellen und die menschliche Gesundheit zu fördern. Das Spezial 510(k)-Programm ist effizient und im Einklang mit dem am wenigsten belastenden Prüfverfahren, das ausländischen Herstellern hilft, ihre Geräte in den USA zu verkaufen und Patienten einen zeitnahen Zugang zu neuen Medizinprodukten zu ermöglichen.
Für weitere Erläuterungen zum speziellen 510(k)-Verfahren der FDA wenden Sie sich an Freyr – einen ausgewiesenen Regulierungsexperten. Bleiben Sie informiert. Bleiben Sie konform.