2 Min. Lesezeit
Shonin (Zulassung vor dem Inverkehrbringen) ist der regulatorische Weg für die Registrierung von Medizinprodukten in Japan. Der Shonin-Weg ist hauptsächlich für die Registrierung von Medizinprodukten der Klassen II und III vorgesehen, für die keine Klassifizierungsstandards der PMDA verfügbar sind. Auch für Hochrisikoprodukte der Klasse IV müssen Hersteller den Shonin-Antrag einreichen. Die PMDA ist für die Prüfung und Genehmigung des Shonin-Antrags zuständig.
Welche anderen Registrierungswege für Medizinprodukte gibt es in Japan?
Neben Shonin werden auch die Wege Todokede und Ninsho für die Zulassung von Medizinprodukten in Japan genutzt. Medizinproduktehersteller können einen dieser Wege wählen, abhängig von der Risikoklasse des Produkts und der Verfügbarkeit von Vergleichsprodukten in Japan. Der Hersteller muss die Klassifizierung des Produkts ermitteln und prüfen, ob der japanische Industriestandard (JIS) verfügbar ist, bevor er den anwendbaren Registrierungsweg festlegt.
- Todokede (Einreichung vor dem Inverkehrbringen) – Dies gilt für Produkte der Klasse I und verlangt von Herstellern, eine Benachrichtigung vor dem Inverkehrbringen bei der PMDA zur Genehmigung einzureichen.
- Ninsho (Zertifizierung vor dem Inverkehrbringen) – Dies gilt für generische Produkte der Klassen II und III, die Zertifizierungsstandards (JIS-Standards) aufweisen. Die registrierte Zertifizierungsstelle (RCB) ist für die Prüfung und Genehmigung des Antrags zuständig.
Was sind die Voraussetzungen für die Shonin-Registrierung?
Hersteller, die ihre Produkte über den Shonin-Weg registrieren, müssen die Einreichungen sorgfältig planen. Sie müssen Folgendes sicherstellen:
- Einreichung allgemeiner Produktdaten, wie z. B. Medizinproduktkategorie, Verwendungszweck, Daten zur Wirksamkeits-Risikoanalyse, klinische Daten usw.
- Bereitstellung der Zusammenfassung der technischen Dokumentation (STED)
- Bereitstellung von Dokumenten ausschließlich in japanischer Sprache
- Die ausländischen Hersteller müssen zwingend einen Marketing Authorization Holder (MAH) oder einen Designated Marketing Authorization Holder (DMAH) benennen.
- Ausländische Hersteller müssen ein Zertifikat für die Registrierung ausländischer Hersteller (FMR) für ihre Produktionsstätten erhalten.
Was sind die QMS-Anforderungen für die Produktregistrierung über den Shonin-Weg?
Hersteller müssen alle QMS-Anforderungen gemäß Verordnung 169 erfüllen. Der Sponsor oder DMAH oder MAH muss sich an die PMDA wenden. Die PMDA führt eine detaillierte QMS-Inspektion der Herstelleranlage durch und stellt bei zufriedenstellender Umsetzung des QMS das Zertifikat aus.
Wie ist der Registrierungsprozess für die Gerätezulassung im Rahmen des Shonin-Verfahrens?
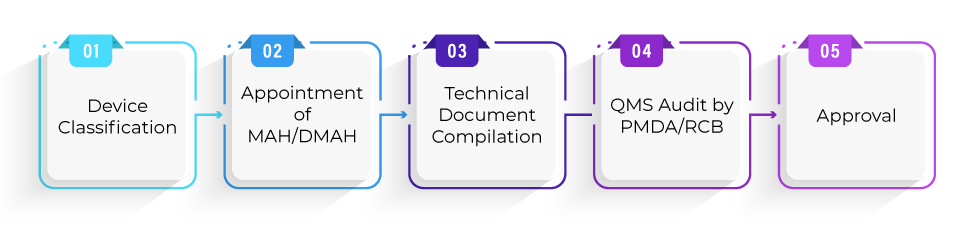
Wie ist der durchschnittliche Zeitrahmen für die Gerätezulassung im Rahmen des Shonin-Verfahrens?
Die PMDA benötigt in der Regel 12 Monate für die technische Bewertung ab dem Datum des Eingangs des Shonin-Antrags. Der Hersteller muss die für die Erstellung der Einreichungsunterlagen oder die Durchführung klinischer Studien erforderliche Zeit in seinen Projektzeitplänen berücksichtigen.
Gibt es eine Ablauffrist für die Geräteregistrierung im Rahmen des Shonin-Verfahrens?
Die Registrierung von Medizinprodukten läuft nicht ab, aber der Sponsor sollte die QMS-Zertifikate alle fünf (05) Jahre erneuern.
Japan ist ein lukrativer Markt, der jedoch von Natur aus regulatorische Komplexitäten und sprachliche Barrieren mit sich bringt. Die Hersteller müssen diese Faktoren berücksichtigen und ihre Markteinführungsstrategie (GTM) für Japan proaktiv planen. Hersteller von Medizinprodukten und IVD können sich dafür entscheiden, alle regulatorischen Feinheiten an einen zuverlässigen regulatorischen Partner auszulagern und die Ressourcen zu nutzen, um sich auf andere wesentliche Komponenten zu konzentrieren.
Um mehr über die Shonin-Zulassung für Medizinprodukte in Japan oder andere PMDA-Vorschriften in Japan zu erfahren, kontaktieren Sie noch heute die Regulierungsexperten von Freyr.