
2 min de lecture
Une demande 510(k) ou une notification préalable à la mise sur le marché est une demande adressée à la Food and Drug Administration (FDAUS visant à démontrer que le dispositif destiné à être commercialisé est sûr et efficace, c'est-à-dire qu'il est substantiellement équivalent à un dispositif déjà commercialisé légalement ou à un dispositif de référence. Voici les trois (03) types de demandes 510(k) qu'un fabricant de dispositifs médicaux peut soumettre :
- Traditionnel
- Abrégé
- 510(k) spécial
Dans cet article, nous examinerons les cas dans lesquels votre demande pourrait relever du deuxième type, à savoir une procédure 510(k) simplifiée, conformément auxFDA US .
Une demande 510(k) abrégée sert à démontrer l'équivalence substantielle par rapport à une norme reconnue, une mesure de contrôle spécifique ou des lignes directrices, au moyen d'une déclaration de conformité (DoC). Dans le cadre d'une demande abrégée, les fabricants démontrent l'équivalence substantielle par rapport à des normes reconnues en s'appuyant sur des documents d'orientation ou des DoC, plutôt que sur un dispositif de référence, afin de faciliter l'examenFDA US . Vous trouverez ci-dessous l'organigramme permettant de déterminer l'équivalence substantielle pour une demande 510(k) abrégée.
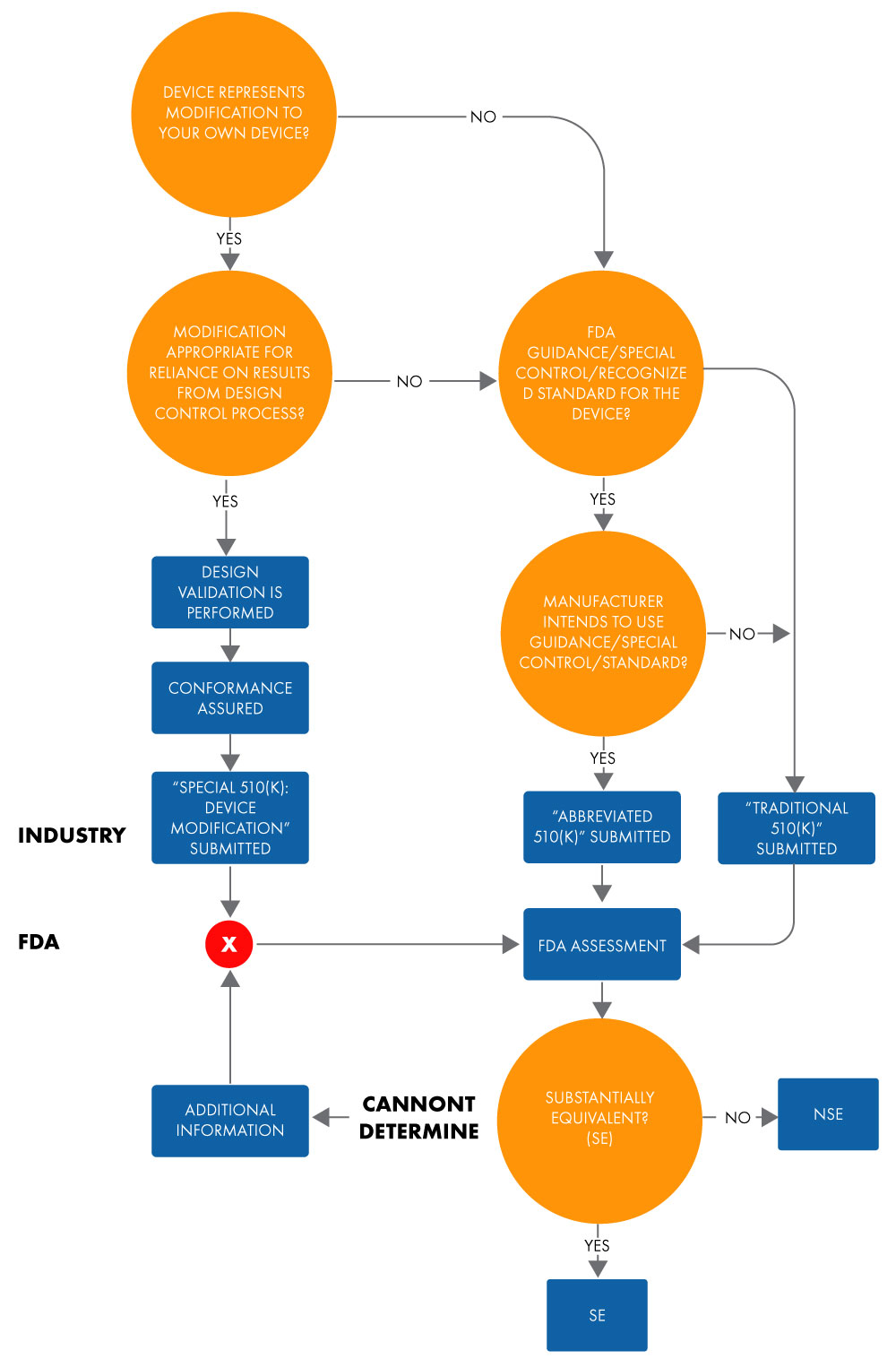
Figure 1 : Intention de commercialiser un dispositif via 510(k)
Le terme « abrégé » suggère que ce type de processus d'approbation 510(k) est plus court. Cependant, ce n'est pas entièrement vrai. Cela prend autant de temps qu'une approbation 510(k) traditionnelle. Il en va de même pour la documentation et les coûts. De plus, le format des 510(k) traditionnels et abrégés, en termes de chapitrage et de structure, est similaire.
Lors de la soumission d'un 510(k) abrégé, vous devez vous baser sur les éléments identifiés dans le 21 CFR 807.87 (soumissions 510[k] traditionnelles). Vous pouvez choisir de soumettre un 510(k) abrégé lorsque la soumission repose sur les éléments suivants :
- DocumentsFDA : lorsdu dépôt d'une demande abrégée 510(k), vous devez joindre un rapport de synthèse décrivant le respect du document d'orientation pertinent et la manière dont celui-ci a été utilisé au cours du développement et des essais du dispositif.
- Démonstration de conformité aux contrôles spéciaux pour le type de dispositif : Vous devez vous conformer aux contrôles spéciaux tels que les normes de performance, la Surveillance après commercialisation (PMS), les registres de patients, l'élaboration et la diffusion de lignes directrices, de recommandations, etc., qui offrent une assurance raisonnable de la sécurité et de l'efficacité du dispositif. Une demande 510(k) abrégée qui s'appuie sur un ou plusieurs contrôles spéciaux doit inclure les éléments suivants. Un rapport sommaire décrivant l'adhésion aux contrôles spéciaux et la manière dont ils ont été utilisés lors du développement et des tests du dispositif.
- Comment les contrôles spéciaux ont été utilisés pour traiter un risque spécifique ou un problème.
- Informations décrivant tout écart par rapport aux contrôles spécifiques et les tentatives du fabricant de s'y conformer.
- Norme(s) de consensus volontaire : Vous êtes tenu de fournir une DoC conforme à la norme reconnue pour une soumission 510(k) abrégée qui s'y appuie. Une DoC doit inclure les éléments suivants :
- Le nom et l'adresse du demandeur/parrain responsable de la DoC.
- Détails de l'identification du produit/dispositif, y compris les codes de produit, le nom commercial du dispositif, le numéro de modèle et toute autre donnée d'identification unique du produit spécifique à la DoC en question.
- Une déclaration de conformité.
- Une liste des normes auxquelles la DoC est applicable, y compris la ou les options sélectionnées pour chaque norme, le cas échéant.
- Le numéroFDA auprès deFDA US pour chaque norme.
- La date et le lieu d'émission de la DoC.
- La signature, le nom imprimé et la fonction du promoteur responsable de la DoC.
- Toute(s) limitation(s) concernant la validité de la DoC (par exemple, la durée de validité de la déclaration, ce qui a été testé, les concessions faites concernant les résultats des tests, etc.)
En conclusion, un 510(k) abrégé est un moyen utile pour les fabricants de dispositifs de démontrer une équivalence substantielle aux normes reconnues ou aux contrôles spéciaux en utilisant une Déclaration de Conformité (DoC). Pour être éligible à un 510(k) abrégé, les fabricants de dispositifs doivent fournir un rapport sommaire expliquant leur adhésion aux documents d'orientation pertinents, démontrer leur conformité aux contrôles spéciaux et fournir des Déclarations de Conformité (DoC) aux normes reconnues. Il est cependant important de noter que le processus d'approbation, la documentation et le coût d'un 510(k) abrégé sont similaires à ceux d'un 510(k) traditionnel.
Votre dispositif médical est-il éligible à une demande 510(k) abrégée ? Pour toute assistance concernant le dépôt de votre demande 510(k) abrégée, contactez notre expert en réglementation. Restez informé ! Restez conforme !









