

3 minuty czytania
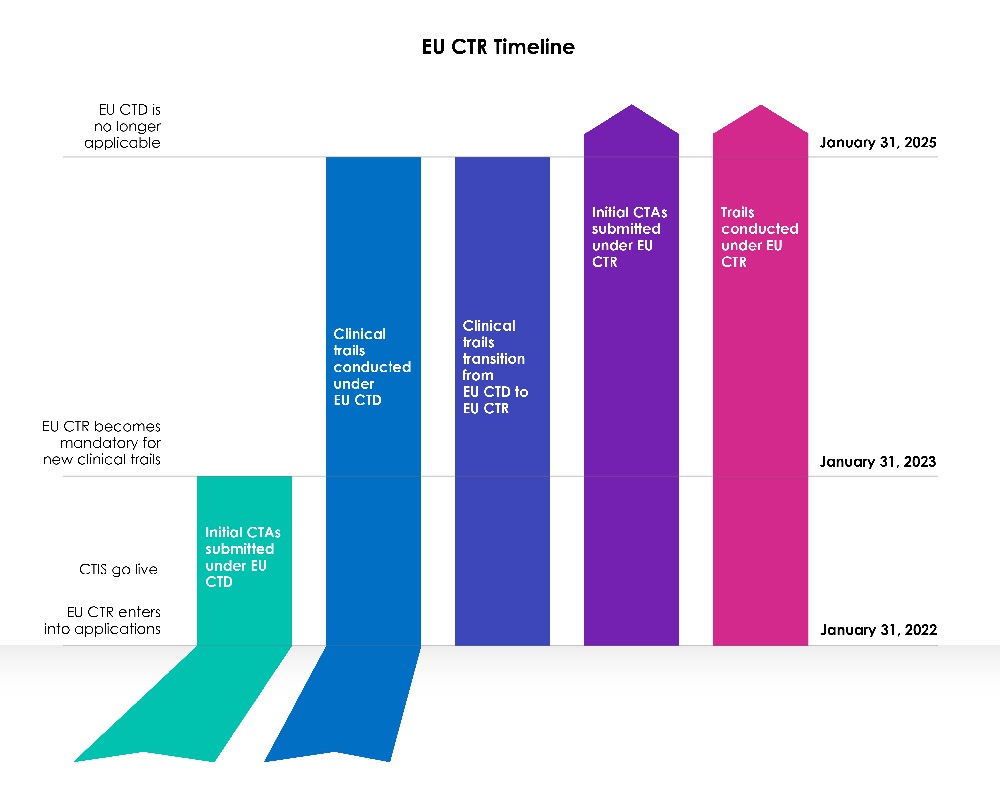
Od 31 stycznia 2022 r. nowe rozporządzenie Unii Europejskiej (UE) w sprawie badań klinicznych (CTR) stało się obowiązkowe, uchylając dyrektywę w sprawie badań klinicznych 2001/20/WE. Rozporządzenie to harmonizuje protokoły oceny i nadzoru nad badaniami klinicznymi w całej UE. Wytyczne zostały zmienione, aby promować jednolite podejście do badań klinicznych, jednocześnie podkreślając bezpieczeństwo uczestników badań klinicznych i zwiększoną jawność.
Rozporządzenie ustanawia nowy dwuczęściowy system oceny dla wszystkich badań klinicznych w UE. Część I polega na naukowej ocenie podstawowej dokumentacji badania klinicznego, a Część II na etycznej ocenie dokumentacji na poziomie krajowym. Po tej dwuczęściowej ocenie każde państwo członkowskie podejmie jednolitą decyzję w sprawie badania i powiadomi sponsora za pośrednictwem Systemu Informacji o Badaniach Klinicznych.
Terminy przejściowe dla nowych wnioskodawców
Trzyletni (03) okres przejściowy rozpoczął się wraz z datą uruchomienia unijnego CTIS.
Rok 1 (31 stycznia 2022 r. – 30 stycznia 2023 r.):
Dyrektywa Unii Europejskiej (UE) w sprawie badań klinicznych 2001/20/WE (EU-CTD) reguluje badania kliniczne w UE od 2004 roku. Dążyła do standaryzacji zasad i znacząco poprawiła bezpieczeństwo pacjentów w badaniach klinicznych. W praktyce jednak doprowadziła do niezamierzonych konsekwencji. W pierwszym roku po wdrożeniu CTIS, sponsorzy mogli wybrać, czy złożyć nowy wniosek o badanie kliniczne (CTA) w ramach Systemu Informacji o Badaniach Klinicznych (CTIS) zgodnie z Dyrektywą w sprawie badań klinicznych (CTD: Dyrektywa 2001/20/WE), czy też korzystać z CTIS zgodnie z obowiązującym prawodawstwem, Rozporządzeniem w sprawie badań klinicznych (EU) nr 536/2014.
Obie koncepcje były wykonalne, a sponsorzy mieli możliwość wyboru, którą ścieżkę legislacyjną obrać.
Member States były gotowe do korzystania z Systemu Informacji o Badaniach Klinicznych (CTIS) i przyjmowały wnioski zgodnie z nowym prawodawstwem, Rozporządzeniem w sprawie Badań Klinicznych (EU CTR), już pierwszego dnia działania CTIS.
Lata 2 i 3 (31 stycznia 2023 r. – 31 stycznia 2025 r.):
Od 31 stycznia 2023 roku wszystkie nowe wnioski o badania kliniczne muszą być składane za pośrednictwem CTIS zgodnie z nowym prawodawstwem (CTR).
Nowe wnioski o badania kliniczne (CT) nie mogą być składane w EudraCT na podstawie Dyrektywy o Badaniach Klinicznych (CTD). Dyrektywa UE o Badaniach Klinicznych nie zezwala już na przyjmowanie nowych Member States po 31 stycznia 2023 r. Badania prowadzone na podstawie CTD muszą najpierw zostać przeniesione, po czym dodatkowy wniosek dotyczący Member State może zostać złożony za pośrednictwem EU CTIS.
Dla Istniejących Wnioskodawców
Wnioski o pozwolenie na badanie kliniczne (CT) złożone przed 30 stycznia 2023 r. na podstawie starych przepisów (CTD) za pośrednictwem EudraCT, będą mogły być prowadzone do zakończenia na podstawie tej Dyrektywy ((CTD: Dyrektywa 2001/20/WE), do 30 stycznia 2025 r. Procedury pozostaną niezmienione, a sponsorzy będą mogli składać istotne modyfikacje i powiadomienia o zakończeniu badania zgodnie z wymogami rozporządzenia. EudraCT pozostanie aktywne w okresie przejściowym, aby umożliwić kontynuację tych badań.
Ważne jest jednak, aby pamiętać, że wnioski przejściowe mogą być składane w dowolnym momencie w ciągu trzyletniego (03) okresu przejściowego, a sponsorzy są zachęcani do ukończenia procesu wystarczająco wcześnie w okresie przejściowym, aby zapewnić ciągłość badań klinicznych w UE po 30 stycznia 2025 r., biorąc pod uwagę dni ustawowo wolne od pracy i dwutygodniowe (02) zimowe wstrzymanie biegu zegara.
Nieprzenoszalne badania
- Badania, które zakończyły się lub zakończą tuż przed końcem okresu przejściowego UE/EOG, nie mogą zostać przeniesione.
- Jeśli powiadomienie o zakończeniu badania zostało zakończone we wszystkich krajach członkowskich UE/EOG, ale globalne zakończenie badania nie zostało jeszcze zgłoszone, badanie nie powinno być przenoszone. Zgodnie z Dyrektywą, globalne zakończenie badania i wyniki podsumowujące badanie powinny zostać opublikowane za pośrednictwem EudraCT.
- Badania rozpoczęte przed wdrożeniem Dyrektywy 2001/20/WE nie korzystają z takiej procedury przejściowej. Jeśli są to badania interwencyjne i muszą być kontynuowane po zakończeniu fazy przejściowej CTR, należy złożyć nowy wniosek o pozwolenie na badanie kliniczne zgodnie z rozporządzeniem CTR.
- Badania pediatryczne prowadzone poza UE/EEA, którym przypisano numer EudraCT, również nie powinny być konwertowane.
- Badań wstrzymanych po zakończeniu okresu przejściowego nie można przenieść. Wznowienie badania w tych okolicznościach wymagałoby złożenia nowego wniosku o pozwolenie na badanie kliniczne zgodnie z rozporządzeniem CTR.
Unijny CTIS regularnie otrzymuje aktualizacje techniczne od EMA w celu poprawy swoich funkcji i działania. Gdy w CTIS wprowadzane są istotne zmiany, EMA publikuje noty wydania, przedstawiające, co zostało zmienione w systemie. Aktualizacje mogą obejmować ulepszenia istniejących funkcji i działania, dodanie nowych funkcji oraz usprawnienia funkcjonalne i techniczne. Doświadczony partner regulacyjny może sprostać potencjalnym wyzwaniom i pomóc sponsorom w przejściu badań dla istniejących i przyszłych badań w ramach strategii rozwoju klinicznego. Kliknij tutaj, aby dowiedzieć się więcej o CTIS i doświadczeniu Freyr w tej dziedzinie: https://www.freyrsolutions.com/medical-devices/regulatory-affairs.









