
3 Min. Lesezeit
In der sich ständig wandelnden und streng regulierten Medizinproduktebranche ist die Gewährleistung von Produktqualität und Patientensicherheit nicht nur eine gesetzliche Verpflichtung, sondern auch eine strategische Notwendigkeit. Wirksame Systeme für das Beschwerdemanagement und die Marktüberwachung gemäß ISO 13485:2016 bilden das Rückgrat jedes soliden Qualitätsmanagementsystems (QMS). Diese Mechanismen gewährleisten die Einhaltung globaler Vorschriften und stärken das Vertrauen der Kunden sowie die Glaubwürdigkeit der Marke.
Lassen Sie uns einen Blick darauf werfen, wie das Beschwerdemanagement und die Wachsamkeit eine grundlegende Rolle bei der Aufrechterhaltung der Qualität und der Einhaltung gesetzlicher Vorschriften in der Medizinproduktebranche spielen – mit Erkenntnissen, die sich an ISO 13485:2016 orientieren und durch globale regulatorische Rahmenbedingungen gestützt werden.
Das Beschwerdemanagement nach ISO 13485:2016 verstehen
ISO 13485:2016 definiert eine Beschwerde als jede schriftliche, mündliche oder elektronische Mitteilung, in der Mängel in Bezug auf die Identität, Qualität, Haltbarkeit, Zuverlässigkeit, Gebrauchstauglichkeit, Sicherheit oder Leistung eines Medizinprodukts geltend gemacht werden. Diese Norm verlangt von Medizinprodukteherstellern, im Rahmen eines wirksamen Qualitätsmanagementsystems dokumentierte Verfahren für die Bearbeitung von Beschwerden festzulegen, einschließlich der Bewertung, Untersuchung und Klärung von Beschwerden.
Ein System zur Bearbeitung von Beschwerden ist entscheidend für die Einhaltung von Vorschriften, die Aufrechterhaltung der Kundenzufriedenheit und die Verbesserung der Gerätequalität. Der Prozess muss strukturiert und nachvollziehbar sein und folgende Schritte umfassen:
zur Entgegennahme und Dokumentation von Beschwerden Alle Beschwerden müssen unverzüglich erfasst werden, wobei möglichst detaillierte Angaben zur Art des Problems, zu den Gerätespezifikationen und zum Modell, zu den Nutzungsbedingungen sowie zu den für den Nutzer oder Patienten eingetretenen Folgen zu machen sind.- Prüfung auf Meldepflicht
Nach ihrer Erfassung müssen Beschwerden geprüft werden, um festzustellen, ob es sich um meldepflichtige Vorfälle handelt. In den USA sind Hersteller von Medizinprodukten beispielsweise verpflichtet, bestimmte Produktprobleme im Rahmen des von der FDA regulierten Medical Device Reporting (MDR)-Systems zu melden. Die Hersteller müssen die geltenden Vorschriften hinsichtlich der Meldefristen einhalten. - Untersuchung und Ursachenanalyse
Bei berechtigten Beschwerden sollte eine eingehende Untersuchung erfolgen. Die Ermittlung der Ursache ist für die Umsetzung wirksamer Korrektur- und Vorbeugungsmaßnahmen (CAPA) von entscheidender Bedeutung. - Korrektur- und Vorbeugungsmaßnahmen (CAPA)
Auf der Grundlage der Ergebnisse der Medizinprodukteprüfung müssen Unternehmen CAPAs einleiten, um sowohl das unmittelbare Problem als auch dessen Wiederauftreten zu beheben und künftige Vorkommnisse zu verhindern. - Rückkopplungsschleife und Abschluss
Sobald das Problem behoben ist, sollte die Beschwerde formell abgeschlossen werden, wobei eine vollständige Dokumentation zu erstellen ist und der Beschwerdeführer gegebenenfalls über den Sachverhalt informiert werden muss, sofern dies für das Medizinprodukt zutrifft.
Wachsamkeit: Eine tragende Säule der Marktüberwachung
Unter Vigilanz versteht man die Überwachung und Meldung von unerwünschten Ereignissen und Vorfällen, sobald ein Medizinprodukt auf dem Markt ist. Gemäß ISO 13485:2016 und den entsprechenden internationalen Vorschriften ist diese kontinuierliche Überwachung unerlässlich für die frühzeitige Erkennung potenzieller Risiken und die Einleitung rechtzeitiger Maßnahmen.
Das im Rahmen der Medizinprodukteverordnung (EU MDR) aktualisierte Überwachungssystem der Europäischen Union verpflichtet Hersteller von Medizinprodukten zur Erstellung von Plänen zur Überwachung nach dem Inverkehrbringen. Diese Pläne müssen Verfahren zur Erhebung und Auswertung von Daten aus der praktischen Anwendung eines Produkts enthalten.
In ähnlicher Weise regelt in Indien die Central Drugs Standard Control Organization CDSCO) die Überwachungs- und Beschwerdeverfahren für Medizinprodukte. Sie fördert die Meldung von unerwünschten Ereignissen und Produktfehlern, um die Sicherheit und Wirksamkeit von Medizinprodukten kontinuierlich zu verbessern.
Zu den wesentlichen Bestandteilen eines soliden Überwachungssystems gehören:
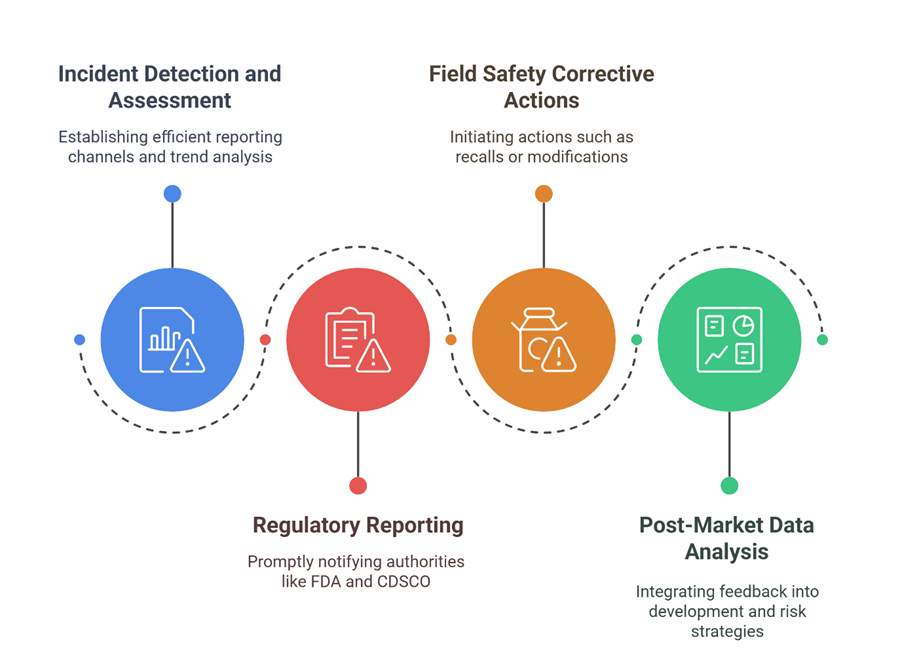
Entwicklung einer integrierten Strategie für Beschwerdemanagement und Arzneimittelüberwachung
Hersteller von Medizinprodukten können ihr Qualitätsmanagementsystem verbessern, indem sie die Bearbeitung von Beschwerden und die Vigilanz in ein einziges, einheitliches System integrieren. Hier sind einige bewährte Verfahren:
Klare Standardarbeitsanweisungen und Rollenbeschreibungen
Standardarbeitsanweisungen (SOPs) sollten die Schritte für die Entgegennahme, Bewertung, Meldung und Klärung von Beschwerden festlegen. Darin wird festgelegt, wer in jeder Phase verantwortlich ist, um Verzögerungen und Lücken zu vermeiden.
- Mitarbeiterschulungen und Sensibilisierungs
en Regelmäßige Mitarbeiterschulungen stellen sicher, dass die Mitarbeiter wissen, wie sie Beschwerden und unerwünschte Ereignisse erkennen und effektiv damit umgehen können. - Digitale Tools und Automatisierung
Der Einsatz von QMS-Software kann die Nachverfolgung von Beschwerden, die Dokumentation und das Berichtswesen optimieren. Die Automatisierung verbessert die Rückverfolgbarkeit und die Konformität mit ISO 13485:2016. - Qualitätskultur und kontinuierliche Verbesserung
Die Förderung einer Philosophie, in der Feedback geschätzt wird, regt zur proaktiven Meldung von Problemen an und fördert Innovationen bei der Qualitätsverbesserung.
Compliance und mehr: Ein Marketingvorteil
Die Einführung von Systemen für das Beschwerdemanagement und die Vigilanz, die ISO 13485:2016 entsprechen, gewährleistet nicht nur die Einhaltung gesetzlicher Vorschriften, sondern positioniert ein Unternehmen auch als vertrauenswürdig und qualitätsorientiert. In einer Branche, in der Sicherheit und Leistung direkten Einfluss auf Menschenleben haben, kann ein solches Engagement ein wirkungsvolles Marketinginstrument sein.
Laut FDA verbessert die Anpassung an internationale Standards den Zugang zu globalen Märkten, stärkt das Vertrauen der Kunden und verringert das Risiko kostspieliger Rückrufaktionen oder Rechtsstreitigkeiten.
| Region | Regulierungsrahmen | Referenznummer/Artikel zur Bearbeitung von Beschwerden | Referenznummer/Artikel für die Vigilanz / Meldung unerwünschter Ereignisse |
| Europäische Union | EU MDR 2017/745 | Artikel 83, Anhang III (PMS-System); Artikel 10 Absatz 9 | Artikel 87–92 (Überwachungssystem), Anhang III |
| Vereinigte Staaten (FDA) | 21 CFR Teil 820 (QSR) | § 820.198 – Beschwerdeakten | 21 CFR Teil 803 – Meldung von Medizinprodukten (MDR) |
| Indien (CDSCO) | Verordnung über Medizinprodukte von 2017 (MDR) | Kapitel VII, Regel 25 und 26 | Kapitel VII, Regel 27 |
| Weltweit (ISO-Norm) | ISO 13485:2016 | Absatz 8.2.2 – Bearbeitung von Beschwerden | Absatz 8.2.3 – Meldungen an die Aufsichtsbehörden |
Fazit
In der heutigen wettbewerbsintensiven Regulierungslandschaft für Medizinprodukte sind ein effektives Beschwerdemanagement und Vigilanzsysteme unverzichtbar. Im Einklang mit ISO 13485:2016 schützen diese Prozesse nicht nur die Patienten, sondern tragen auch zum Aufbau einer widerstandsfähigen, reputationsorientierten Marke bei. Durch Investitionen in systematische Verfahren, kontinuierliche Schulungen und Vigilanzmaßnahmen nach dem Inverkehrbringen können Hersteller regulatorische Anforderungen in Chancen für Wachstum und Marktführerschaft verwandeln.









