
Przegląd usług w zakresie zapewnienia zgodności z unijnym rozporządzeniem IVDR
Rozporządzenie UE w sprawie wyrobów medycznych do diagnostyki in vitro (EU IVDR) stanowi nową podstawę prawną dla wprowadzania do obrotu na rynku europejskim wyrobów medycznych do diagnostyki in vitro. Rozporządzenie IVDR UE ma zastąpić obecną dyrektywę UE w sprawie wyrobów medycznych do diagnostyki in vitro (UE 98/79/WE). Jako rozporządzenie europejskie będzie miało zastosowanie we wszystkich państwach członkowskich UE oraz państwach członkowskich Europejskiego Stowarzyszenia Wolnego Handlu (EFTA), bez konieczności transponowania go do prawa krajowego tych państw. Jako sprawdzony partner w zakresie regulacji, firma Freyr oferuje producentom usługi w zakresie zgodności i doradztwa dotyczącego IVDR w UE, aby zapewnić zgodność z przepisami dotyczącymi diagnostyki in vitro i oznakowania CE.
Umów się na spotkanie z naszymi ekspertami ds. unijnego rozporządzenia IVDR
Rozporządzenie IVDR – harmonogram przejścia
Nowe rozporządzenie europejskie IVDR, które ma wejść w życie 26 maja 2022 r., jest postrzegane jako istotna zmiana w zakresie nadzoru regulacyjnego nad wyrobami medycznymi do diagnostyki in vitro. Ogólny zarys procesu przejścia na IVDR przedstawia się następująco –
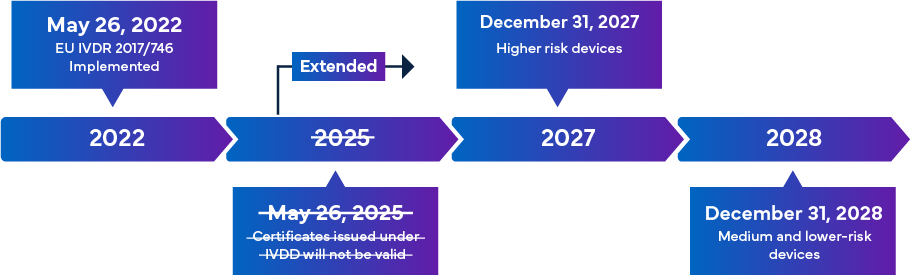
Po pełnym wdrożeniu przepisy dotyczące zgodności z rozporządzeniem IVDR gwarantują, że niemal wszystkie wyroby do diagnostyki in vitro wprowadzane na rynek europejski podlegają ocenie przez jednostkę notyfikowaną (ON) zgodnie z rozporządzeniem IVDR oraz certyfikacji oznakowania CE w ramach procedury zatwierdzania wyrobów do diagnostyki in vitro. W tym scenariuszu, oprócz dostosowania się do zaktualizowanej klasyfikacji IVDR, producenci muszą natychmiast zweryfikować kluczową dokumentację techniczną, aby pomyślnie przejść rejestrację wyrobu do diagnostyki in vitro (IVD) i uzyskać oznakowanie CE. Wymagania IVDR różnią się w zależności od klasy ryzyka wyrobu do diagnostyki in vitro (IVD). Ogólnie rzecz biorąc, w celu certyfikacji wyrobów do diagnostyki in vitro (IVD) producenci muszą wykonać następujące czynności:
- Analiza dokumentacji technicznej zgodnie z rozporządzeniem IVDR (rozporządzenie UE 2017/746)
- Przygotowanie raportu z oceny wyników (PER) dla wszystkich wyrobów do diagnostyki in vitro
- Monitorowanie działania wyrobu po wprowadzeniu do obrotu (PMPF) zgodnie z załącznikiem XIII część B do rozporządzenia IVDR.
- Raport z monitoringu zgodny z art. 82 rozporządzenia IVDR
Freyr wspiera swoich klientów w przeprowadzaniu systematycznego przeglądu literatury naukowej oraz pomaga im w planowaniu i przygotowaniu planu oceny ryzyka (PER) w celu zapewnienia zgodności z przepisami dotyczącymi wyrobów do diagnostyki in vitro. Freyr dysponuje wyspecjalizowanymi ekspertami, którzy świadczą usługi doradcze w zakresie IVDR oraz zapewniają wsparcie w zakresie nadzoru po wprowadzeniu do obrotu (SPM), stanowiącego integralną część systemu zarządzania jakością producenta, a także monitorowania wyników po wprowadzeniu do obrotu (SPPM).
Skorzystaj z porad ekspertów dotyczących zapewnienia zgodności z unijnym rozporządzeniem IVDR
Usługi w zakresie zgodności z unijnym rozporządzeniem IVDR – DOŚWIADCZENIE I KORZYŚCI
- Plan przejściowy dotyczący zgodności z rozporządzeniem IVDR
- Analiza techniczna i ocena luk w wymaganiach IVDR dotyczących GSPR (ogólnych wymagań dotyczących bezpieczeństwa i działania)
- Wspieranie przygotowania dokumentacji technicznej zgodnie z wymogami rozporządzenia IVDR
- Raporty dotyczące naukowej zasadności oparte na literaturze przedmiotu i/lub danych wewnętrznych
- Raporty dotyczące wyników klinicznych oparte na literaturze przedmiotu i/lub danych wewnętrznych
- Dane kliniczne lub raporty z oceny wydajności
- Protokoły i raporty dotyczące monitorowania wyników po wprowadzeniu do obrotu (SPPM)
- Protokoły i sprawozdania z nadzoru po wprowadzeniu do obrotu (SPM)
- Opracowywanie/weryfikacja innych dokumentów, takich jak ulotki dołączane do opakowań/instrukcje użytkowania (IFU), skrócone instrukcje (QRI), instrukcje obsługi/eksploatacji itp.

- Zapewnienie zgodności z rozporządzeniem IVDR, rejestracji IVD oraz oznakowania CE
- Dogłębna znajomość przepisów oraz doświadczenie w kluczowych obszarach objętych unijnym rozporządzeniem IVDR
- Model realizacji oparty na zarządzaniu projektami, mający na celu zapewnienie dotrzymania harmonogramu.
- Wewnętrzni eksperci ON (ocena raportu przez interaktywnych recenzentów ON)
- Zespoły specjalistyczne posiadające wszechstronną wiedzę w kluczowych obszarach oraz w zakresie konkretnych rodzajów rozwiązań
- Wkład ekspertów ds. wyrobów medycznych z różnych dziedzin w zapewnienie zgodności z wymogami
- Kompleksowe usługi w zakresie zgodności, audytu i planowania
- Duże doświadczenie w zapewnianiu spójności wyników pracy (terminowość i jakość)
