2 minuty czytania
Głównym celem US FDA jest nieustanne badanie i wypełnianie luki między procesami regulacyjnymi, aby zapewnić nieprzerwany import i sprzedaż nowych, wysokiej jakości wyrobów medycznych na rynku US. W 1998 roku US FDA opublikowała program zatytułowany “Nowy Paradygmat 510(k): Alternatywne Podejścia do Wykazania Zasadniczej Równoważności w Zgłoszeniach Przedrynkowych.” Ma on na celu ustanowienie wydajnej ścieżki składania wniosków FDA 510(k), która uwzględnia pewne zmiany w już zatwierdzonym wniosku 510(k). To nowe zgłoszenie 510(k) oferuje (03) trzy rodzaje zgłoszeń, a mianowicie: specjalne 510(k), skrócone 510(k) i tradycyjne 510(k). W 2019 roku US FDA wydała dokument wytycznych dotyczący specjalnego 510(k), opisujący opcjonalną ścieżkę dla producentów, którzy wprowadzają pewne dobrze zdefiniowane modyfikacje do swoich legalnie wprowadzonych do obrotu wyrobów.
Dlaczego specjalne zgłoszenie 510(k)?
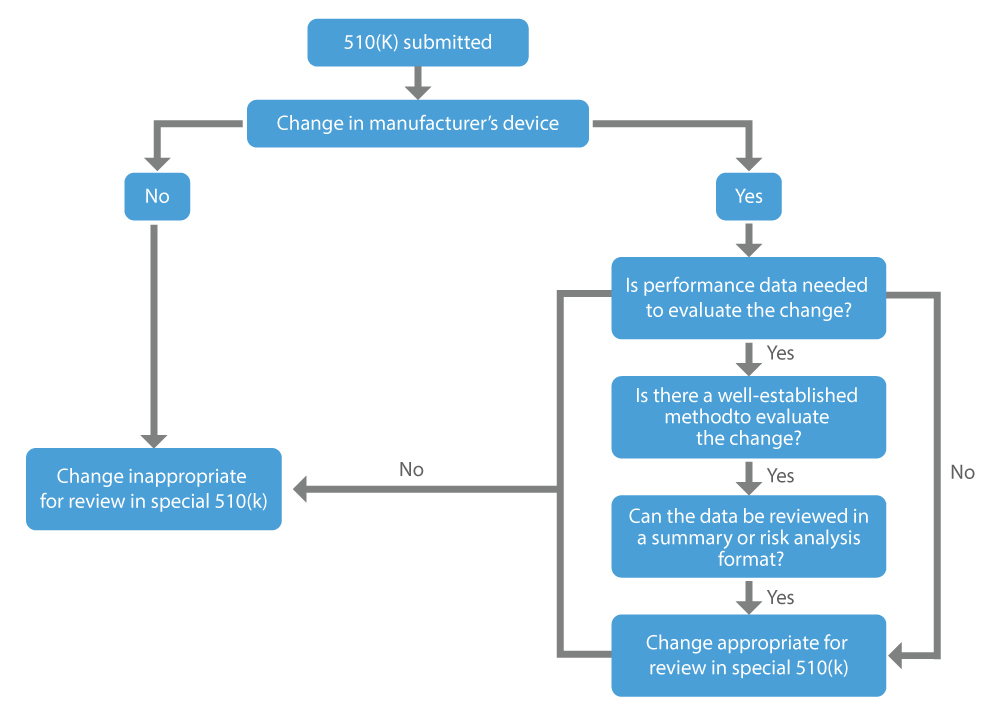
Gdy producent chce uzyskać zatwierdzenie modyfikacji, które wprowadził w już wprowadzonym na rynek wyrobie, czyli w istniejącym wyrobie, może złożyć wniosek o specjalne 510(k). Główne czynniki, które należy wziąć pod uwagę przy określaniu, czy zmiana w istniejącym wyrobie może być odpowiednia dla specjalnego 510(k), są następujące:
- Zmiana dotyczy legalnie wprowadzonego do obrotu wyrobu referencyjnego, należącego do zgłaszającego.
- Dane dotyczące wydajności nie są wymagane, lub dostępne są dobrze ugruntowane metody, jeśli uznano to za konieczne do oceny zmiany.
- Wszystkie dane dotyczące wydajności wspierające ustalenie istotnej równoważności mogą być przeglądane w formie podsumowania lub analizy ryzyka.
Wymagane dokumenty dla specjalnego 510(k)
- List przewodni
- Nazwa legalnie wprowadzonego na rynek (istniejącego) wyrobu producenta oraz numer 510(k)
- Szczegółowy opis zmian wprowadzonych w wyrobie, które doprowadziły do złożenia nowego wniosku 510(k)
- Porównanie zmodyfikowanego wyrobu z wyrobem dopuszczonym do obrotu w formie tabelarycznej
- Inne zmiany w oznakowaniu lub projekcie
- Zwięzłe podsumowanie działań kontroli projektowania
- Na podstawie analizy ryzyka, identyfikacja działań weryfikacyjnych i/lub walidacyjnych wymaganych do zachowania zgodności z 21 CFR 820.30
- Formularz wskazań do stosowania
- Oświadczenie, że zgłaszający przestrzegał i obecnie nie narusza wymogów procedury kontroli projektu określonych w 21 CFR 820.30, a dokumentacja jest dostępna do wglądu na żądanie
Specjalny harmonogram przeglądu 510(k) przez US FDA
Zgodnie z wytycznymi FDA „Polityka odmowy przyjęcia dla zgłoszeń 510(k)”, termin przeglądu specjalnych zgłoszeń 510(k) wynosi trzydzieści (30) dni od daty ich otrzymania.
Kiedy ubiegać się o specjalne 510(k)?
US FDA stale dąży do zapewnienia bezpiecznych i skutecznych wyrobów medycznych w celu promowania zdrowia ludzi. Program Special 510(k) jest efektywny i zgodny z procedurą oceny o najmniejszym obciążeniu, co pomaga zagranicznym producentom sprzedawać swoje wyroby w USA i umożliwia pacjentom terminowy dostęp do nowych wyrobów medycznych.
W celu uzyskania dalszych wyjaśnień dotyczących specjalnego procesu 510(k) FDA, skontaktuj się z Freyr – sprawdzonym ekspertem regulacyjnym. Bądź na bieżąco. Zachowaj zgodność.