2 Min. Lesezeit
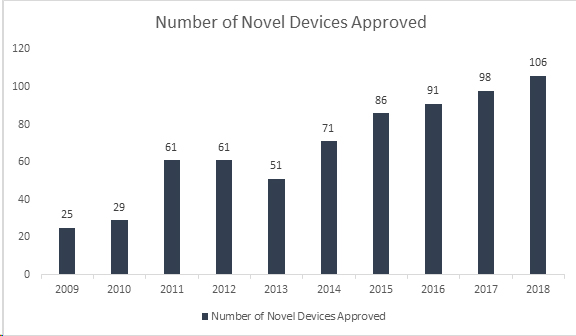
Wussten Sie schon? Die US Food and Drug Administration (FDA) hat 2018 einen weiteren Rekord aufgestellt, indem sie 106 neuartige Geräte zugelassen hat, was es zum erfolgreichsten Jahr für Medizinprodukte macht. Mit dieser Leistung hat die FDA ihren 2017 aufgestellten 40 Jahre alten Rekord von 99 zugelassenen neuartigen Geräten übertroffen und zeigt damit ein kontinuierliches Wachstum über die letzten 8 Jahre. Zu den zugelassenen Geräten gehören eine Reihe innovativer Produkte wie automatisierte Insulindosierungssysteme für Kinder, die kleinste Herzklappe der Welt für Neugeborene, die erste mobile medizinische App zur Unterstützung bei der Bewältigung von Opioid- und Drogenmissbrauchsstörungen sowie Technologien der künstlichen Intelligenz, die diabetische Retinopathie diagnostizieren.
Die FDA hat stets die Sicherheit und Innovation von Medizinprodukten gefördert, um deren hohe Qualität zu gewährleisten. Um mit der steigenden Zahl neuartiger Gerätegenehmigungen und deren Sicherheit Schritt zu halten, plant die FDA, den Zulassungsweg für Medizinprodukte zu „modernisieren“. Nach Angaben der Behörde könnte die Modernisierung der Zulassung eine neue Befugnis erfordern. Im Jahr 2018 veröffentlichten die FDA und das Center for Devices and Radiological Health (CDRH) ein gemeinsames Dokument, in dem festgestellt wurde, dass 510(k) eine der beiden Einreichungsarten war, die der Definition neuartiger Geräte hinzugefügt wurden. Zusammen damit wurden auch die Humanitarian Device Exemptions (HDEs) in die Definition neuartiger Geräte aufgenommen, nachdem Änderungen am Breakthrough Medical Device Program des CDRH aufgrund des 21. Century Act vorgenommen wurden.
Der Vorschlag der FDA und des CDRH ist eine Reaktion auf die potenzielle Notwendigkeit, neue Befugnisse zur Modernisierung des 510(k)-Verfahrens zu verfolgen. Das Motiv des Vorschlags ist es, die Verwendung von Prädikatsprodukten, die als im Wesentlichen gleichwertig (SE) gelten und älter als 10 Jahre sind, zu begrenzen, um Innovationen zu fördern. Dies ist ein Fortschritt im Hinblick auf die im April 2018 von der Behörde veröffentlichte Leitlinie. Der Entwurf wurde von der FDA veröffentlicht, um die Erweiterung des verkürzten 510(k)-Programms beim CDRH der FDA unter dem Titel „Sicherheits- und leistungsbasierter Wegweiser“ vorzuschlagen. Er wurde eingeführt, um die Belastung durch Bestimmungen für Medizinprodukte zu verringern. Der Ansatz zielt auch darauf ab, die Effizienz der Überprüfung von 510(k)-Einreichungen zu erhöhen und somit den Druck auf die Behörde zu reduzieren.
Hier sind einige der wichtigsten Punkte der Leitlinie:
- Das neue Verfahren bewertet die Sicherheit und Wirksamkeit der Produkte anhand festgelegter Sicherheits- und Leistungsstandards.
- Trotz der neuen Standards müssen die Produkte den bestehenden Standards entsprechen, um vermarktet werden zu können.
- Moderne Technologie wird anhand moderner Standards getestet.
- Dieser Ansatz wird den Wettbewerb bei der Entwicklung sichererer Produkte fördern.
Die Anzahl der Medizinprodukte, die zur Zulassung eingereicht werden, ist im Laufe der Jahre exponentiell gestiegen. Dies hat der FDA einen weiten Spielraum gegeben, innovative Maßnahmen zur Verbesserung der Zulassungswege zu ergreifen und umzusetzen. Die Behörde ist fest von den Vorzügen dieses Vorschlags überzeugt, doch die Reaktion der Industrie muss noch entschlüsselt werden.
Da die FDA ständig neue Leitfäden zur Aktualisierung der Medizinprodukte-Registrierung veröffentlicht, ist es für Hersteller von Medizinprodukten notwendig, diese im Auge zu behalten und entsprechend zu handeln. Bleiben Sie auf dem Laufenden. Bleiben Sie konform.









