
Lesezeit: 5 Min.
Medizinproduktesoftware in Südkorea wird zur Diagnose, Behandlung und Überwachung von Patienten im modernen Gesundheitssystem eingesetzt. Sie umfasst sowohl eingebettete Software, die in Medizinprodukte integriert ist, als auch eigenständige Software, die auf PCs, mobilen Geräten und webbasierten Diensten genutzt werden kann. Das Ministerium für Lebensmittel- und Arzneimittelsicherheit (MFDS) in Südkorea ist für die Regulierung von Medizinproduktesoftware und die Gewährleistung ihrer Sicherheit und Wirksamkeit zuständig. Am 05. Juli 2023 legte das MFDS Kriterien für die Zulassung und Prüfung von Medizinproduktesoftware fest; diese Vorschriften bieten eine Struktur, der zivile Antragsteller folgen können, wenn sie Software zur Zulassung oder Überprüfung einreichen.
Die Vorschriften behandeln eine Vielzahl von Themen, darunter den Anwendungsbereich, Anforderungen an die technische Dokumentation und Berichte zur Konformitätsprüfung. Zusätzlich zu den MFDS-Leitlinien gibt es internationale Standards und Leitlinien, die für Medizinproduktesoftware gelten, wie den Standard der International Electrotechnical Commission (IEC) 62304 für Software-Lebenszyklusprozesse und die Leitlinien der United States Food and Drug Administration (US FDA) für mobile medizinische Anwendungen.
Software-Entwicklungsplan und Anforderungsanalyse
- Der Softwareentwicklungsplan beschreibt den gesamten Ansatz zur Softwareentwicklung, einschließlich Spezifikationen, Methoden und Entwicklungswerkzeugen. Er umfasst auch die Verifizierung, das Risikomanagement für Medizinprodukte, das Konfigurationsmanagement und die Dokumentation.
- Die Anforderungsanalyse legt die Anforderungen an die Software von Medizinprodukten fest, einschließlich Risikokontrollmaßnahmen und Verifizierungsmethoden. Durch sorgfältige Planung und Analyse des Softwareentwicklungsprozesses können Entwickler sicherstellen, dass die resultierende Software die notwendigen Standards für Sicherheit und Wirksamkeit erfüllt.
- Der Verifizierungsbericht zur Software-Konformität enthält eine Gliederung des Softwareentwicklungsplans, die Dokumentenkontrollnummer des Herstellers und eine Übersicht über die Anforderungsanalyse. Durch die Einhaltung dieser Richtlinien kann Software für Medizinprodukte mit Zuversicht entwickelt werden, im Wissen, dass sie strengen Tests unterzogen wurde und die notwendigen Standards für Sicherheit und Wirksamkeit erfüllt.
Verifizierung und Validierung von Software für Medizinprodukte
- Die Verifizierung von Software für Medizinprodukte stellt sicher, dass die Software die festgelegten Anforderungen erfüllt.
- Die Validierung von Software für Medizinprodukte stellt sicher, dass die Software die Bedürfnisse des Benutzers und die beabsichtigte(n) Verwendung(en) erfüllt.
- Der Bericht zur Verifizierung und Validierung von Medizinproduktesoftware beschreibt den Verifizierungs- und Validierungsprozess, einschließlich des Produktnamens, der Version und der Namen der Personen, die den Bericht geprüft und genehmigt haben. Der Bericht kann je nach den Eigenschaften der Software variieren, sollte aber eine Beschreibung der Software, die verwendeten Verifizierungs- und Validierungsmethoden sowie die Testergebnisse enthalten.
Betriebsumgebung und Software unbekannter Herkunft (SOUP)
- Wenn die Software von spezifischer Hardware abhängig ist, wie z. B. eingebettete Software, sollte das technische Dokument die Hardwarespezifikationen beschreiben.
- Wenn die Software jedoch eigenständig ist und für den Betrieb auf allgemeiner Hardware entwickelt wurde, muss die Betriebsumgebung in der Dokumentation beschrieben werden. Dies umfasst die empfohlenen Mindestspezifikationen, wie z. B. Microsoft Windows 10 oder höher.
- Wenn die Software für Medizinprodukte zusätzlich kommerzielle Software unbekannter Herkunft (SOUP) enthält, muss eine Betriebsumgebung geschaffen werden, um die ordnungsgemäße Funktion zu gewährleisten. Durch die sorgfältige Beschreibung der Betriebsumgebung und die Berücksichtigung jeglicher SOUP können Entwickler sicherstellen, dass ihre Software für Medizinprodukte sicher und wirksam für den beabsichtigten Gebrauch ist.
Risikomanagement und Dokumentationsanforderungen für Medizinprodukte
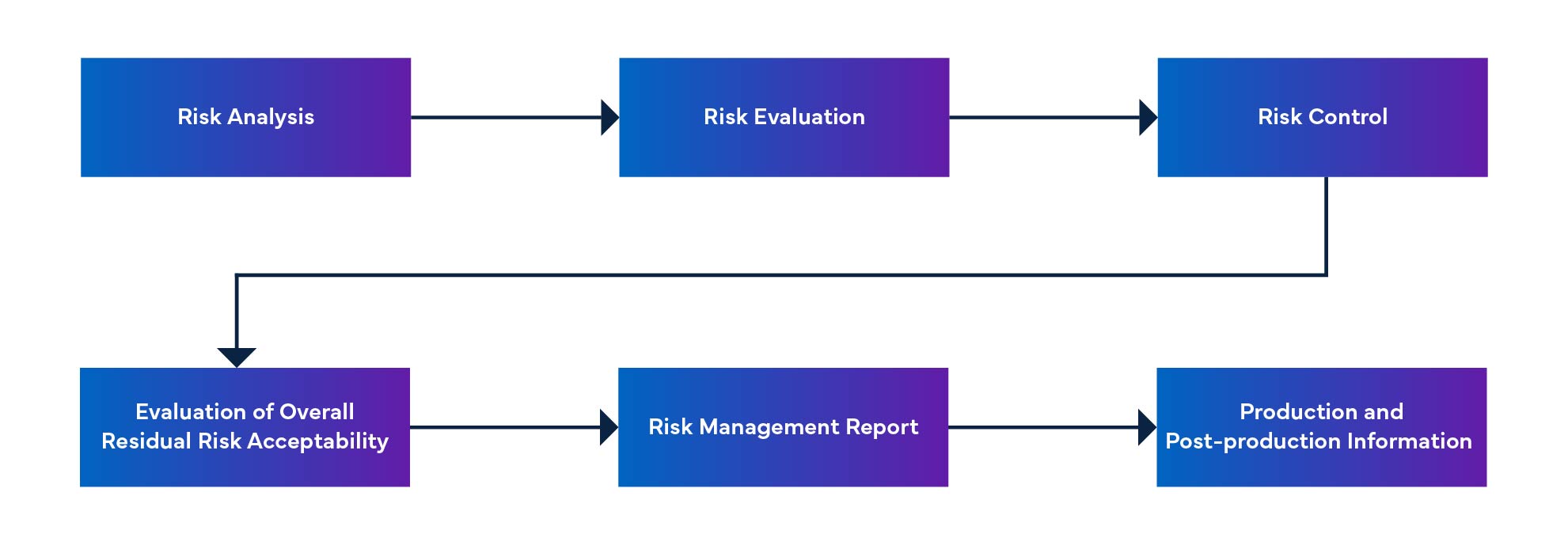
- Der Risikomanagementprozess für Software als Medizinprodukt umfasst die Identifizierung gefährlicher Situationen, die Festlegung von Risikokontrollmaßnahmen, die Überprüfung dieser Maßnahmen und die Verwaltung von Softwareänderungen.
- Das MFDS-RM-Dokument zum Softwarerisikomanagement liefert Informationen zum Softwarerisikomanagement.
- Zusätzlich sind Dokumentationsanforderungen unerlässlich, um sicherzustellen, dass die Software die notwendigen Standards für Sicherheit und Wirksamkeit erfüllt.
- Der Softwareentwicklungsplan, die Anforderungsanalyse für Software von Medizinprodukten sowie die Verifizierungs- und Validierungsberichte der Software müssen in der Dokumentation enthalten sein.
- Der Verifizierungsbericht zur Software-Konformität skizziert die Dokumentationsanforderungen; er enthält auch eine Gliederung der anwendbaren Dokumente und die Dokumentenkontrollnummer des Herstellers.
Abbildung 1: Risikomanagementprozess für Medizinprodukte
Ungelöste Anomalien und Korrekturmaßnahmen für SaMD-Software
- Das MFDS-PR-Dokument (Software-Problembehebung) skizziert den Prozess zur Software-Problembehebung, der Problemmeldung, Analyse, Implementierung und Verifizierung umfasst.
- Das Dokument enthält auch eine Liste ungelöster Probleme, Bugs, Defekte und Anomalien sowie eine Restrisikobewertung für das Softwaresystem.
- Die zur Behebung dieser Probleme ergriffenen Korrekturmaßnahmen müssen im Software-Wartungsplan dokumentiert werden, der gemäß dem Software-Wartungsprozess erstellt wird.
- Das MFDS-Wartungsdokument enthält Informationen zur Wartung und Fehlerbehebung von SaMD-Software.
Überprüfung technischer Dokumente und Einreichungsanforderungen für SaMD-Software
Die wichtigsten Prüfdokumente während des Prüfprozesses sind Leistungsdaten, der Konformitätsbestätigungsbericht und Verifizierungs- und Validierungsdaten der Software für Medizinprodukte, die Software-Design-Spezifikation (SDS), die Software-Anforderungsspezifikation (SRS) für Medizinprodukte sowie Verifizierungs- und Validierungsberichte. Der Konformitätsbestätigungsbericht und der Verifizierungs- und Validierungsbericht der Software für Medizinprodukte müssen eingereicht werden.
Risikomanagement für Software von Medizinprodukten
- Identifizierung potenzieller Gefahren im Zusammenhang mit der Software und ihrer Nutzung.
- Bewertung der Schwere der Risiken im Zusammenhang mit diesen Gefahren.
- Implementierung von Risikokontrollmaßnahmen, um die Wahrscheinlichkeit von Schäden zu minimieren.
- Überwachung und Überprüfung der Wirksamkeit dieser Risikokontrollmaßnahmen.
- Dokumentation aller Risikomanagementaktivitäten und -entscheidungen für Medizinprodukte.
In einem Softwaresystem werden Softwareelemente in kleinere Teile, einschließlich detaillierter Softwareelemente, unterteilt. Wenn ein Element nicht weiter unterteilt werden kann, wird es als Einheit bezeichnet. Das System ermöglicht die Unterteilung bis auf die Einheitsebene und hilft so, die Sicherheitsstufe für jedes Softwareelement zu bestimmen. Durch das Zusammenfügen dieser Softwareelemente können wir die Sicherheitsstufe für das gesamte Softwaresystem ermitteln.
Abbildung 2: Zerlegung und Integration von Medizinproduktesoftware
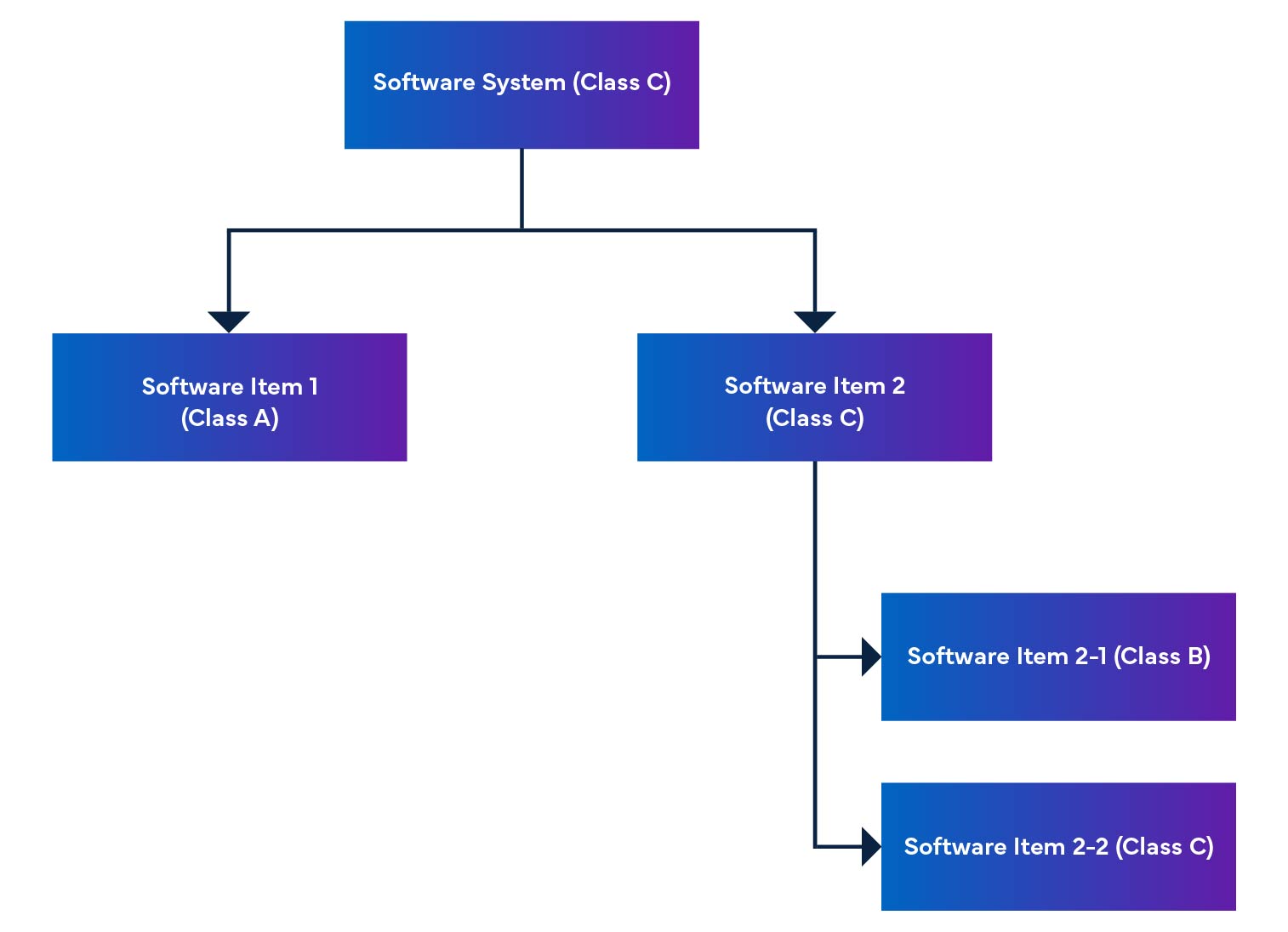
Die Verordnung erwähnt auch die Software-Sicherheitsbewertung, die eine Bewertung zur Identifizierung der Risiken von SaMD-Software ist (siehe Tabelle 1).
Tabelle 1: Definition der Sicherheitsbewertung
| Einstufung | Definition der Sicherheitsklasse für Medizinproduktesoftware |
| Klasse A | Keine Möglichkeit von Verletzungen oder körperlichem Schaden. |
| Klasse B | Weniger schwerwiegende Verletzungen (leichte Verletzungen) sind wahrscheinlich. |
| Klasse C | Möglichkeit schwerer Verletzungen oder des Todes. |
Software-Konfigurationsmanagement
- Pflege einer genauen und aktuellen Dokumentation für alle Softwareversionen, Änderungen und Aktualisierungen.
- Sicherstellung, dass die gesamte Dokumentation ordnungsgemäß geprüft und genehmigt wird.
- Implementierung von Verfahren zur Verwaltung von Software-Konfigurationsänderungen.
- Dokumentation aller Aktivitäten und Entscheidungen im Software-Konfigurationsmanagement.
Softwarewartung
- Regelmäßiges Testen und Überwachen der Software, um sicherzustellen, dass sie für ihren vorgesehenen Verwendungszweck sicher und effektiv bleibt.
- Implementierung von Verfahren zur Behebung auftretender Probleme, einschließlich Fehlerbehebungen und Software-Updates.
- Dokumentation aller Software-Wartungsaktivitäten und Entscheidungen.
Fehlerbehebung
- Ermittlung der Grundursache des Problems.
- Implementierung von Korrekturmaßnahmen zur Behebung des Problems.
- Dokumentation des gesamten Fehlerbehebungsprozesses für zukünftige Referenzzwecke.
Durch Befolgen der oben genannten Richtlinien können Entwickler sicherstellen, dass alle Probleme mit ihrer Medizinproduktesoftware ordnungsgemäß behoben und dokumentiert werden und dass die Software die notwendigen Anforderungen für die Zulassung oder Prüfung erfüllt.
Wenn Sie ein Medizinproduktehersteller sind, der die Einhaltung der südkoreanischen Standards für Medizinproduktesoftware anstrebt, können die Regulierungsexperten von Freyr Sie durch die komplexe Regulierungslandschaft des Landes führen. Wir stellen sicher, dass Ihre Produkte den neuesten südkoreanischen Medizinproduktevorschriften entsprechen, um eine reibungslose Konformität zu gewährleisten. Kontaktieren Sie uns, um mehr zu erfahren!









