3 Min. Lesezeit
Im Laufe der Jahre hat sich mit den Fortschritten in Software und Digitalisierung die Art und Weise, wie Medizinprodukte verwaltet und bereitgestellt werden, grundlegend verändert. Die Integration von Software in Medizinprodukte hat rapide zugenommen und treibt unglaubliche Fortschritte bei der Bereitstellung von Gesundheitslösungen in verschiedenen Bereichen voran, wie Diagnose, Krankheitsprävention und Behandlung von Verletzungen oder Krankheiten.
Der Einfluss von Software auf die Sicherheit und Leistung von Medizinprodukten war jedoch fraglich, insbesondere wenn das Produkt selbst nur aus Software besteht. Daher werden die Vorschriften für Medizinproduktesoftware ständig überarbeitet, um festzulegen, wann Software als Medizinprodukt (SaMD) gilt. Kürzlich hat der Beirat der Europäischen Kommission – die Medical Device Coordination Group (MDCG) – sich darauf konzentriert, die Vorschriften für Medizinproduktesoftware zu verbessern, und einen Leitfaden veröffentlicht, der den anzuwendenden Ansatz beschreibt, um festzustellen, ob eine Software ein Medizinprodukt ist oder nicht. Was beschreibt dieser Leitfaden? Wir enthüllen es.
Der Geltungsbereich des Leitfadens
Der MDCG-Leitfaden umfasst sowohl Medizinproduktesoftware als auch In-vitro-Diagnostika (IVD)-Medizinproduktesoftware. Gemäß dem Dokument ist eine Medizinproduktesoftware (MDSW) als Software definiert, die allein oder in Kombination für einen Zweck verwendet werden soll, wie er in der Definition eines „Medizinprodukts“ in der Medizinprodukte-Verordnung 2017/745 (MDR) oder der In-vitro-Diagnostika-Verordnung 2017/746 (IVDR) festgelegt ist. Er legt die Kriterien fest, die anzuwenden sind, um zu bestimmen, ob eine zu prüfende Software ein Medizinprodukt ist oder nicht, und beabsichtigt, zusätzliche Klarstellungen und Empfehlungen zur MDSW für Medizinproduktehersteller und andere Parteien bereitzustellen.
Zunächst legt der Leitfaden die wichtigsten Begriffe fest, die im Zusammenhang mit MDSW verwendet werden, darunter:
Zweckbestimmung: Die Verwendung, für die ein Produkt gemäß den vom Hersteller auf dem Etikett, in der Gebrauchsanweisung oder in Werbe- oder Verkaufsmaterialien oder -aussagen bereitgestellten Daten und wie vom Hersteller in der klinischen Bewertung angegeben, bestimmt ist.
Zubehör: Ein Artikel, der, obwohl er selbst kein Medizinprodukt ist, vom Hersteller dazu bestimmt ist, zusammen mit einem oder mehreren Medizinprodukten verwendet zu werden, um die Medizinprodukte spezifisch in Übereinstimmung mit ihrer/ihren Zweckbestimmung(en) zu ermöglichen oder die Funktionalität der Medizinprodukte spezifisch und direkt im Hinblick auf ihre/ihren Zweckbestimmung(en) zu unterstützen. Zusätzlich erwähnt die MDCG, dass das Software-Zubehör die Verwendung eines Medizinprodukts steuern oder beeinflussen kann und die vom Hersteller bereitgestellten Gebrauchsanweisungen und andere Dokumentationen Details zur Auswahl der geeigneten Software und des Zubehörs enthalten sollten.
Software: Sie steht für eine Reihe von Anweisungen, die Eingabedaten verarbeitet und Ausgabedaten erzeugt.
Bestimmung der Medizinproduktesoftware
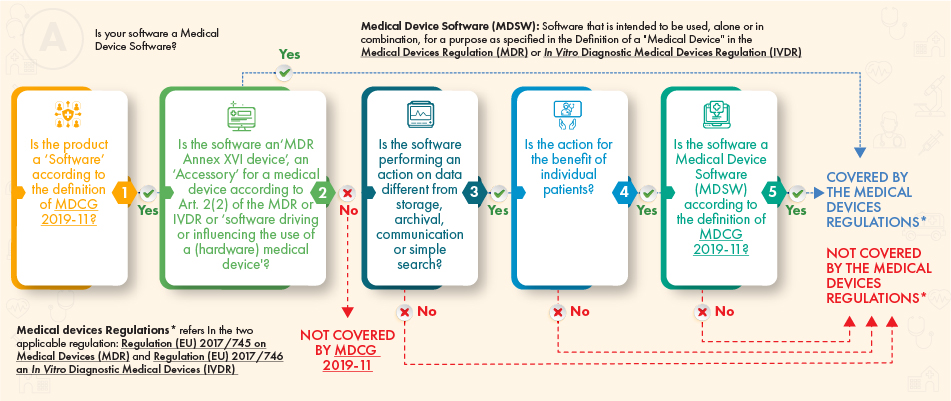
Gemäß dem oben genannten Leitfaden-Flussdiagramm sollte die betreffende Software der Regulierung unterliegen, wenn sie die folgenden Kriterien erfüllt:
- Die Definition eines Medizinprodukts, ein Zubehör dazu, oder die Funktionen des Medizinprodukts steuert, oder
- Sie führt eine zusätzliche Datenverarbeitung durch (nicht nur Speicherung oder Kommunikation) und ihre Aktion schafft Vorteile für die Patienten und erfüllt die Definition von Medizinproduktesoftware gemäß dem MDCG-Leitfaden.
Bestimmung von In-vitro-Diagnostika-Software
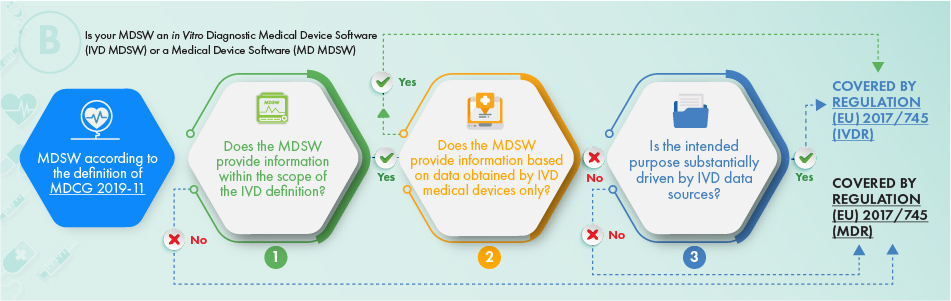
Das obige Flussdiagramm beschreibt den anzuwendenden Ansatz in Bezug auf Produkte, die für In-vitro-Diagnosezwecke bestimmt sind. Um festzustellen, ob die betreffende Software der Regulierung unterliegen sollte, sind die folgenden Kriterien zu berücksichtigen:
- Die Definition eines Medizinprodukts, ein Zubehör dazu, oder die Funktionen des Medizinprodukts steuert, oder
- Sie liefert die Informationen, die üblicherweise von In-vitro-Diagnostika bereitgestellt werden, und nur die Informationen, die von einem In-vitro-Diagnostikum gesammelt wurden, oder
- Die Zweckbestimmung der Software bezieht sich auf IVDR-Angelegenheiten.
Gemäß dem MDCG-Leitfaden beeinflusst die Art der Verbindung zwischen der Medizinproduktesoftware und dem Produkt nicht die Qualifizierung der Software als Produkt gemäß MDR und IVDR. Eine Medizinproduktesoftware kann entweder als eigenständiges Produkt existieren oder in ein Hardwareprodukt integriert sein und präzisiert die folgenden regulatorischen Anforderungen:
- Unter Berücksichtigung ihrer Qualifizierung und Klassifizierung muss ein eigenständiges Medizinproduktesoftwareprodukt dem vollen Umfang der regulatorischen Verfahren gemäß der geltenden Gesetzgebung unterzogen werden.
- Eine Medizinproduktesoftware, die ein integraler Bestandteil oder Teil eines Hardware-Medizinprodukts ist, könnte im Rahmen des vereinfachten Verfahrens auf den Markt gebracht werden. Sie würde nicht separat, sondern während der allgemeinen Bewertung des Hardware-Medizinprodukts selbst überprüft werden.
Zusammenfassend deckt der MDCG-Leitfaden die wesentlichen Aspekte der Klassifizierung von Medizinproduktesoftware und die Festlegung der anzuwendenden regulatorischen Anforderungen ab. Medizinproduktehersteller, Softwareentwickler und andere Beteiligte müssen die MDCG-Empfehlungen befolgen und umsetzen, um die Einhaltung der Vorschriften zu gewährleisten. Um weitere Einblicke in die Einstufung Ihrer Software als Medizinprodukt zu erhalten, konsultieren Sie einen Regulierungsexperten. Bleiben Sie informiert. Bleiben Sie konform.









