2 minuty czytania
Shonin (Zatwierdzenie przed wprowadzeniem do obrotu) to ścieżka regulacyjna dla rejestracji wyrobów medycznych w Japonii. Ścieżka Shonin jest przeznaczona głównie do rejestracji wyrobów medycznych klasy II i III, dla których nie są dostępne standardy klasyfikacji PMDA. Również w przypadku wyrobów wysokiego ryzyka klasy IV, producenci powinni złożyć wniosek Shonin. PMDA jest odpowiedzialna za przegląd i zatwierdzenie wniosku Shonin.
Jakie są inne ścieżki rejestracji wyrobów w Japonii?
Oprócz Shonin, ścieżki Todokede i Ninsho są również wykorzystywane do zatwierdzania wyrobów medycznych w Japonii. Producenci wyrobów medycznych mogą wybrać jedną z nich w zależności od klasy ryzyka urządzenia i dostępności produktów referencyjnych w Japonii. Producent powinien określić klasyfikację urządzenia i zbadać dostępność Japońskiego Standardu Przemysłowego (JIS) przed ustaleniem odpowiedniej ścieżki rejestracji.
- Todokede (Zgłoszenie przed wprowadzeniem na rynek) – Ma zastosowanie do wyrobów klasy I i wymaga od producentów złożenia powiadomienia przed wprowadzeniem na rynek do PMDA w celu uzyskania zatwierdzenia.
- Ninsho (Certyfikacja przed wprowadzeniem na rynek) - Ma zastosowanie do generycznych wyrobów klasy II i III posiadających standardy certyfikacji (normy JIS). Zarejestrowana jednostka certyfikująca (RCB) jest odpowiedzialna za przegląd i zatwierdzenie wniosku.
Jakie są warunki wstępne dla rejestracji Shonin?
Producenci rejestrujący swoje wyroby za pośrednictwem ścieżki Shonin muszą skrupulatnie zaplanować zgłoszenia. Muszą zapewnić co następuje:
- Przesyłanie ogólnych danych dotyczących wyrobu, takich jak kategoria wyrobu medycznego, przeznaczenie, dane z analizy ryzyka skuteczności, dane kliniczne itp.
- Sporządzanie Podsumowania Dokumentacji Technicznej (STED)
- Dostarczanie dokumentów wyłącznie w języku japońskim
- Zagraniczni producenci obowiązkowo muszą wyznaczyć Posiadacza Pozwolenia na dopuszczenie do obrotu produktu leczniczego (MAH) lub wyznaczonego Posiadacza Pozwolenia na dopuszczenie do obrotu produktu leczniczego (DMAH)
- Zagraniczni producenci muszą uzyskać certyfikat Rejestracji Zagranicznego Producenta (FMR) dla swoich zakładów produkcyjnych.
Jakie są wymagania QMS dla rejestracji wyrobów w ramach ścieżki Shonin?
Producenci muszą przestrzegać wszystkich wymagań QMS określonych w Rozporządzeniu 169. Sponsor, DMAH lub MAH musi złożyć wniosek do PMDA. PMDA przeprowadza szczegółową inspekcję QMS w zakładzie producenta i wydaje certyfikat po zadowalającym wdrożeniu QMS.
Jaki jest proces rejestracji w celu zatwierdzenia wyrobu w ramach ścieżki Shonin?
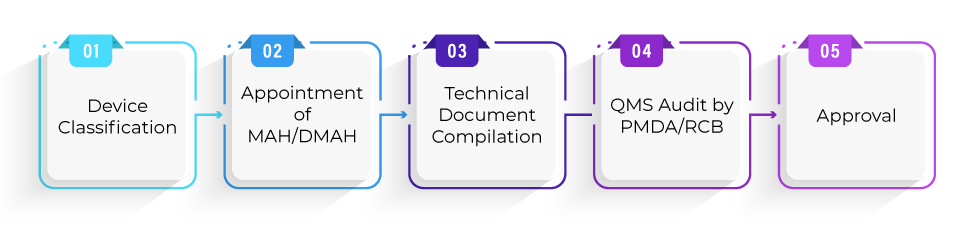
Jaki jest średni czas wymagany do zatwierdzenia wyrobu w ramach ścieżki Shonin?
PMDA zazwyczaj potrzebuje 12 miesięcy na ocenę techniczną od daty otrzymania wniosku Shonin. Producent musi uwzględnić czas potrzebny na przygotowanie dokumentów zgłoszeniowych lub przeprowadzenie badań klinicznych w harmonogramie swojego projektu.
Czy istnieje termin ważności dla rejestracji wyrobu medycznego w ramach ścieżki Shonin?
Rejestracja wyrobu medycznego nie wygasa, ale sponsor powinien odnawiać certyfikaty QMS co pięć (05) lat.
Japonia to lukratywny rynek, ale z natury wiąże się ze złożonościami regulacyjnymi i barierami językowymi. Producenci muszą wziąć pod uwagę te czynniki i proaktywnie zaplanować swoją strategię wejścia na rynek (GTM) w Japonii. Producenci wyrobów medycznych i IVD mogą zdecydować się na zlecenie wszystkich niuansów regulacyjnych wiarygodnemu partnerowi regulacyjnemu i wykorzystać zasoby, aby skupić się na innych istotnych elementach.
Aby dowiedzieć się więcej o zatwierdzeniu wyrobów medycznych Shonin w Japonii lub innych przepisach PMDA Japan, skontaktuj się z ekspertami regulacyjnymi Freyr już dziś.