
Relatório de avaliação clínica (CER) para dispositivos médicos Visão Geral
Qualquer dispositivo destinado a ser comercializado na União Europeia (UE) deve ostentar a marcação CE. De acordo com o EU MDR relativo aos dispositivos médicos, os requisitos para um Relatório de Avaliação Clínica (CER), incluindo os requisitos relativos ao processo e aos dados, variam consoante a classe do dispositivo e são necessários para a obtenção da certificação CE dos dispositivos médicos. Os dispositivos de baixo risco da classe I podem realizar a autocertificação CE. Em contrapartida, outras classes de dispositivos (IIa, IIb, III) devem processar a certificação da marcação CE através de um Organismo Notificado (ON) acreditado. O fabricante deve apresentar o dossiê técnico CE ao ON para avaliação e aprovação da marcação CE, bem como para a emissão do certificado CE. O Relatório de Avaliação Clínica (CER) para dispositivos médicos deve ser apresentado juntamente com o Dossiê Técnico CE para cumprir os requisitos de marcação CE. O CER deve ser atualizado continuamente com novas informações provenientes de verificações de segurança, estudos de acompanhamento e gestão de riscos.
O Relatório de Avaliação Clínica (CER) para dispositivos médicos é um dos relatórios que deve ser submetido juntamente com o Ficheiro Técnico CE para cumprir os requisitos do CER.
O que é um Relatório de Avaliação Clínica (CER)?
A elaboração do relatório de avaliação clínica inclui a avaliação e análise de dados clínicos relacionados com um Dispositivo Médico para verificar a sua segurança e desempenho clínicos. A avaliação clínica de dispositivos médicos baseia-se na análise abrangente de dados clínicos pré e pós-comercialização relevantes para a utilização prevista. O Relatório de Avaliação Clínica inclui dados específicos do dispositivo, bem como quaisquer dados relativos a dispositivos declarados como equivalentes pelo fabricante.
Um relatório de avaliação clínica consiste em literatura científica e dados clínicos analisados, recolhidos quer a partir de um ensaio clínico do seu dispositivo, quer a partir dos resultados de outros estudos sobre dispositivos substancialmente equivalentes, sendo que os fabricantes devem ter acesso total aos dados técnicos, biológicos e clínicos do dispositivo equivalente e demonstrar claramente de que forma o seu dispositivo corresponde a todos os aspetos críticos. O CER de um dispositivo médico demonstra que o dispositivo cumpre a sua finalidade prevista sem expor os utilizadores e os doentes a riscos adicionais.
EU MDR deve ser atualizado anualmente. A frequência das atualizações do CER é determinada com base no risco e depende do dispositivo em questão. Se o dispositivo apresentar riscos significativos, a atualização deve ser feita pelo menos uma vez por ano; caso o dispositivo seja comercializado há um período significativo e esteja bem estabelecido no mercado, o CER pode ser atualizado a cada 2 a 5 anos. Quaisquer alterações introduzidas no projeto do dispositivo e quaisquer novas informações provenientes dos dados de pós-comercialização (PMS) podem exigir uma atualização do Relatório CER.
A avaliação clínica de dispositivos médicos, conforme enquadrado no Relatório de Avaliação Clínica (CER), baseia-se nos fatores listados abaixo.
- Literatura científica atualmente disponível; e/ou
- Investigações clínicas realizadas; ou
- Se a demonstração de conformidade com os requisitos essenciais com base em dados clínicos não for considerada apropriada.
Etapas da Elaboração do Relatório de Avaliação Clínica (CER)
Referindo-nos ao novo Regulamento da UE sobre Dispositivos Médicos (MDR) – 2017/745, existem quatro (04) fases diferentes para realizar uma avaliação clínica de Dispositivos Médicos para preparar um Relatório de Avaliação Clínica (CER) abrangente do MDR da UE.
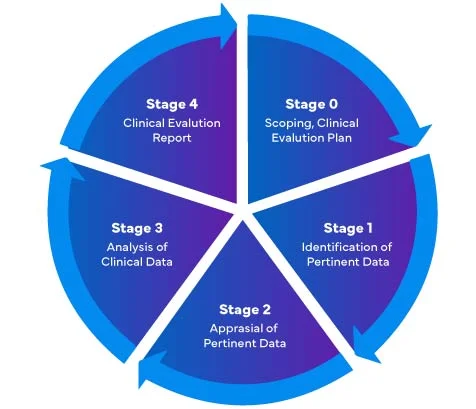
Os fabricantes de Dispositivos Médicos que entram pela primeira vez no mercado da UE devem garantir que o seu Relatório de Avaliação Clínica está em conformidade com os regulamentos EU MDR.
A Freyr oferece serviços End-to-End de Certificação CE a fabricantes de dispositivos, incluindo a elaboração de Relatórios de Avaliação Clínica em conformidade com os regulamentos EU MDR 2017/745 recentemente implementados. Com uma forte experiência regional em Dispositivos Médicos da UE, a Freyr atende aos requisitos específicos de cada agência e personaliza o Relatório de Avaliação Clínica em conformidade.
Obtenha aconselhamento especializado sobre a sua submissão CER
Relatório de Avaliação Clínica (CER)
- Apoio End-to-End à redação de Relatórios de Avaliação Clínica, incluindo pesquisa da literatura, de acordo com a MEDDEV 2.7/1 revisão 4 e as diretrizes do Regulamento Europeu de Dispositivos Médicos (MDR).
- Elaborar um plano de avaliação clínica para a sua organização.
- Identificar, pesquisar, analisar e compilar a literatura científica aplicável e apropriada.
- Desenvolver um modelo de Relatório de Avaliação Clínica para a sua organização.
- Análise de Lacunas para Relatórios de Avaliação Clínica existentes.
- Ferramenta DMS para a sua equipa contribuir coletivamente para a elaboração do Relatório de Avaliação Clínica.
- Integração de Dados de PMS.
- Desenvolver um procedimento operacional padrão para a sua equipa compilar dados de PMS para atualizar os Relatórios de Avaliação Clínica.
- Gestão de atualizações periódicas de Relatórios de Avaliação Clínica existentes, de acordo com as diretrizes do MDR da UE.
- Suporte de dados de PMS para dispositivos existentes no mercado.
- Conformidade com a Marcação CE e serviços de Marcação CE.

- Conformidade assegurada com os regulamentos aplicáveis mais recentes.
- Equipa de especialistas clínicos qualificados.
- Contributos multifuncionais de especialistas em Dispositivos Médicos para cumprir os requisitos.
- Serviço de âmbito completo desde a conformidade, revisão e planeamento.

Perguntas Frequentes (FAQs)
01. O que é um Relatório de Avaliação Clínica (CER) e por que é importante?
Um Relatório de Avaliação Clínica (CER) é um documento científico regulamentar que avalia sistematicamente toda a evidência clínica disponível para demonstrar que um dispositivo médico é seguro, funciona conforme previsto e apresenta resultados aceitáveis em termos de relação benefício-risco para a sua utilização prevista, nos termos EU MDR. Desempenha um papel central na avaliação da conformidade e na conformidade contínua.
02. Quando deve um CER ser elaborado e atualizado?
A elaboração do CER tem início numa fase inicial do desenvolvimento do produto, como parte de um processo de avaliação clínica planeado, e deve ser atualizada regularmente ao longo do ciclo de vida do dispositivo sempre que surjam novas evidências clínicas, dados pós-comercialização ou alterações no perfil de risco, garantindo que a avaliação da relação benefício-risco se mantenha atualizada.
03. Que elementos essenciais deve incluir um CER válido?
Um CER sólido deve refletir uma avaliação estruturada dos dados clínicos relevantes, comparações com as tecnologias mais avançadas, uma análise da relação benefício-risco, contributos da vigilância pós-comercialização e conclusões objetivas sobre a conformidade com os requisitos EU MDR e desempenho EU MDR .
04. De que forma o «estado da arte» influencia um Relatório de Avaliação Clínica?
O «estado da arte» é o ponto de referência da prática clínica e da tecnologia aceites, com o qual se comparam as evidências clínicas; garante que os benefícios e riscos do dispositivo sejam contextualizados em relação aos padrões atuais da medicina, orientando uma interpretação significativa das evidências.
05. É necessário um CER para todos os dispositivos médicos ao abrigo EU MDR?
Sim, os CER são obrigatórios para todos os dispositivos médicos comercializados na UE ao abrigo do MDR, independentemente da classe de risco, uma vez que documentam as evidências clínicas essenciais para demonstrar a conformidade com as expectativas regulamentares em matéria de segurança e desempenho.
06. O que distingue um CER de alta qualidade de um relatório de conformidade básico?
Um CER de alta qualidade integra uma metodologia abrangente de pesquisa bibliográfica, afirmações clínicas claras e alinhadas com a utilização prevista, uma avaliação rigorosa dos dados e análises ponderadas sobre o desempenho clínico e os riscos, indo além do simples cumprimento de requisitos para refletir uma compreensão profunda das expectativas regulamentares e do contexto clínico.
07. Por que razão a Freyr é considerada um parceiro de referência em serviços de Relatórios de Avaliação Clínica (CER)?
A Freyr é amplamente reconhecida pela sua abordagem centrada na regulamentação no desenvolvimento de CER, combinando EU MDR profundo EU MDR , uma avaliação rigorosa das evidências clínicas e estratégias orientadas para o ciclo de vida do produto. As suas equipas multidisciplinares integram conhecimentos clínicos, regulamentares e pós-comercialização para produzir CER cientificamente rigorosos, capazes de resistir ao escrutínio dos organismos notificados e de garantir a conformidade a longo prazo.







