
Por que razão a vigilância pós-comercialização é importante no ciclo de vida dos dispositivos médicos
A Vigilância Pós-Comercialização (PMS) é um elemento fundamental do ciclo de vida dos dispositivos médicos, especialmente no contexto de regulamentações em constante evolução, como EU MDR, o IVDR e os requisitos FDA . À medida que os dispositivos se tornam mais avançados, abrangendo SaMD, dispositivos vestíveis e tecnologias conectadas, o monitoramento contínuo no mundo real e a deteção proativa de riscos são essenciais. Tendências do setor, como evidência do mundo real (RWE), vigilância digital e resultados relatados pelos pacientes, destacam a necessidade de uma avaliação contínua da segurança, desempenho e benefício clínico dos dispositivos. Como parte Vigilância Pós-Comercialização modernas Vigilância Pós-Comercialização , os fabricantes devem incorporar cada vez mais relatórios de tendências, deteção de sinais e avaliações baseadas no risco.

Apesar da sua importância, muitos fabricantes enfrentam desafios para cumprir as expectativas do PMS. Fontes de dados fragmentadas, obrigações de comunicação globais inconsistentes, canais de reclamação multilingues e um escrutínio regulatório cada vez maior criam complexidade operacional. A preparação de planos de PMS, PMSR, PSUR e PMCF requer conhecimentos especializados, interpretação precisa dos dados e coordenação interfuncional. Lacunas na análise de tendências, deteção de sinais, Avaliação de Riscos para a Saúde (HHE) ou relatórios de vigilância de dispositivos médicos podem conduzir a conclusões de auditorias, potenciais recolhas de produtos ou riscos para a continuidade do produto.
A Freyr responde a estes desafios com soluções end-to-end abrangentes e end-to-end , concebidas para satisfazer os requisitos regulamentares globais. As nossas equipas combinam conhecimentos especializados em matéria de regulamentação, metodologias estruturadas e apoio multilingue para otimizar o tratamento de reclamações, a comunicação de vigilância e a documentação de PMS. Com experiência comprovada em SaMD das Classes I a III, IVD e SaMD , a Freyr garante relatórios de alta qualidade, conformidade atempada e preparação para auditorias, tornando-nos um parceiro de confiança para fabricantes de dispositivos médicos que procuram uma gestão fiável e eficiente do ciclo de vida do PMS.
Principais componentes da vigilância pós-comercialização dos dispositivos médicos
Uma vigilância pós-comercialização (PMS) eficaz combina várias atividades interligadas que monitorizam a segurança do dispositivo, o desempenho clínico, a experiência do utilizador e os riscos emergentes ao longo de todo o ciclo de vida comercial do produto. Estes componentes constituem a base das expectativas regulamentares globais ao abrigo EU MDR e do IVDR EU MDR, bem como dos requisitos FDA . Compreender cada um destes elementos é essencial para manter a conformidade, melhorar o desempenho dos dispositivos na prática e mitigar de forma proativa as preocupações em matéria de segurança.
![]()
Gestão de reclamações e eventos adversos
Recebimento, investigação e análise de tendências estruturados das reclamações, com o objetivo de detetar sinais precoces de segurança, garantir o encaminhamento atempado e manter a conformidade global com os requisitos FDA, EU MDR e das normas regionais de notificação de vigilância.![]()
Vigilância e Notificação ao
Identificação, avaliação e comunicação atempadas de eventos adversos e incidentes graves às autoridades reguladoras globais, garantindo uma supervisão contínua da segurança e o cumprimento dos requisitos EU MDR FDA e EU MDR .![]()
Retiradas, correções e exclusões
End-to-end das ações corretivas no terreno, incluindo avaliação de riscos, avaliação de riscos para a saúde (HHE), notificações regulamentares, comunicação e verificações de eficácia, com vista a salvaguardar a segurança dos doentes e proteger a credibilidade do mercado.![]()
Plano de Gestão de Serviços de Manutenção (
, PMSP)
Um plano estruturado de vigilância pós-comercialização que defina responsabilidades, fontes de dados, processos e critérios de avaliação, a fim de garantir um acompanhamento consistente e proativo do desempenho do dispositivo ao longo de todo o seu ciclo de vida.![]()
PMSR, PSUR
ePMCF
Relatórios exigidos pela regulamentação que resumem os dados pós-comercialização, incluindo o relatório de vigilância pós-comercialização (PMSR), atualizações sobre a relação benefício-risco e atividades de acompanhamento clínico, com o objetivo de demonstrar a segurança e o desempenho contínuos do dispositivo.![]()
Relatórios de tendências e evidências do mundo real
Análise dos padrões de reclamações e dos dados de desempenho reais para identificar riscos emergentes, apoiar ações preventivas e melhorar a fiabilidade dos produtos através de práticas estruturadas de elaboração de relatórios de tendências.






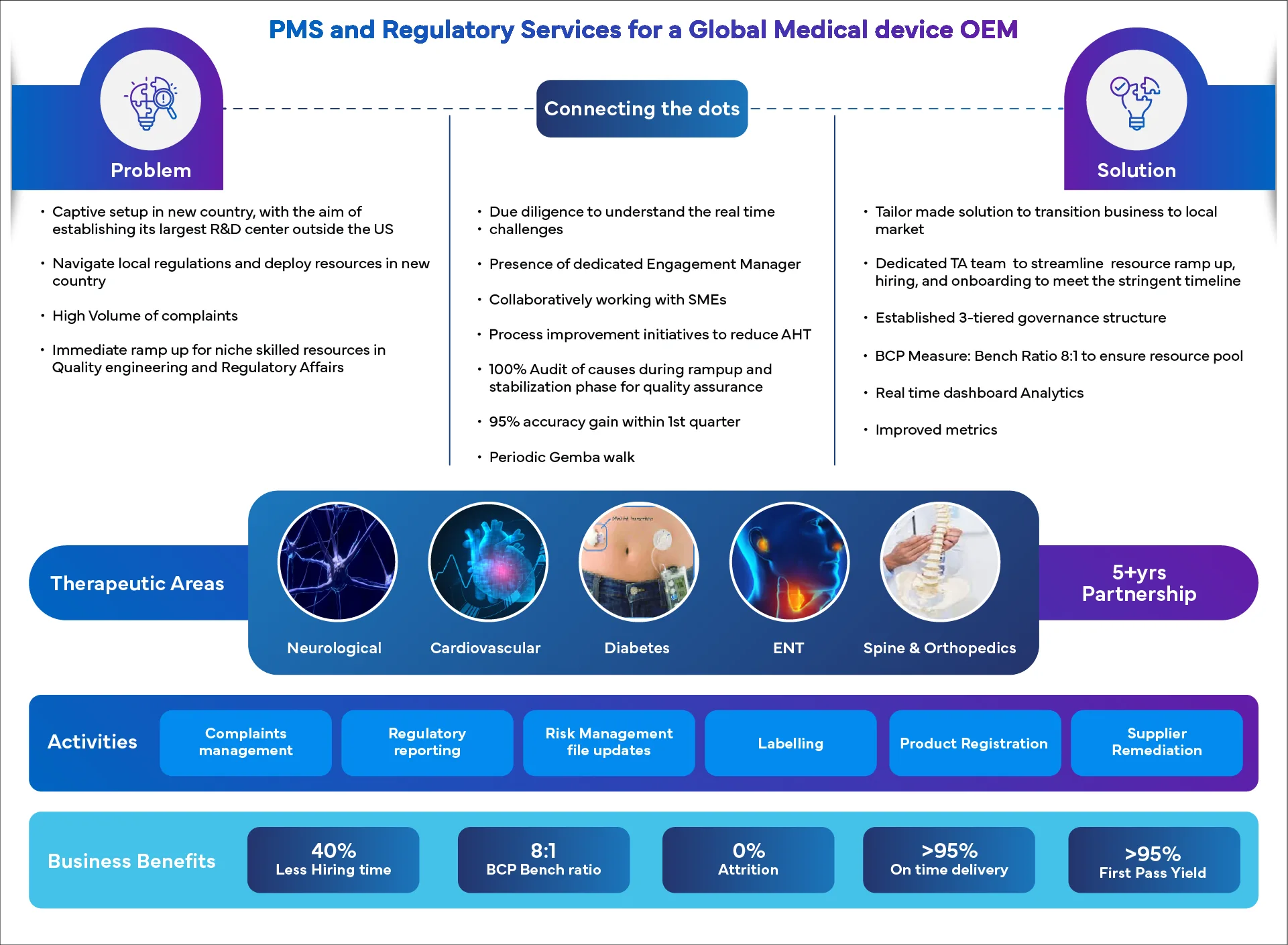
Serviços de Vigilância Pós-Comercialização da Freyr
Agende uma reunião com os nossos especialistas hoje
Em destaque: Resultados reais, histórias reais
Porquê colaborar com a Freyr?
- A vasta experiência nas estruturas regulamentares FDA, EU MDR, do IVDR e do PMS da APAC garante uma conformidade consistente e preparada para inspeções, bem como um forte alinhamento regulamentar nos mercados globais.
- End-to-end , incluindo o tratamento de reclamações, a comunicação de vigilância, os relatórios PMSR e PSUR e PMCF todo o ciclo de vida pós-comercialização seja gerido com precisão e eficiência.
- A análise baseada em IA, a identificação automatizada de tendências e os insights sobre evidências do mundo real ajudam a detetar precocemente riscos emergentes, apoiam a gestão de CAPA e permitem tomar decisões proativas em matéria de segurança.
- Os especialistas em idiomas locais simplificam a triagem, a documentação e o envio de reclamações, garantindo operações do PMS fluidas e precisas em todas as regiões.
- Sucesso comprovado na orientação de fabricantes em auditorias FDA, EU MDR e de organismos notificados, com redução do número de não conformidades, melhores resultados em termos de qualidade e maior confiança na conformidade.
Perguntas Frequentes
01. O que é a vigilância pós-comercialização (PMS) no domínio dos dispositivos médicos e por que razão é importante?
A vigilância pós-comercialização é um processo contínuo e sistemático destinado a monitorizar a segurança, o desempenho e a eficácia na prática dos dispositivos médicos após a sua entrada no mercado. É essencial porque permite aos fabricantes detetar riscos emergentes, validar os benefícios clínicos ao longo do tempo, cumprir as regulamentações globais em constante evolução e tomar decisões baseadas em dados concretos para melhorar a qualidade dos produtos e a segurança dos doentes.
02. Quais são os principais requisitos relativos aos sistemas de gestão de produtos (PMS) ao abrigo EU MDR do IVDR EU MDR ?
EU MDR IVDR EU MDR exigem que os fabricantes mantenham um plano de vigilância pós-comercialização (PMS), elaborem relatórios PMS ou PSURs regulares, realizem PMCF quando necessário e estabeleçam sistemas de notificação de vigilância e análise de tendências. Os regulamentos enfatizam a recolha proativa de evidências, a integração de dados clínicos e a avaliação contínua da relação benefício-risco ao longo de todo o ciclo de vida do dispositivo.
03. Como é que o PMS se integra com a gestão de riscos e a norma ISO 14971?
O PMS integra informações do mundo real diretamente no quadro de gestão de riscos da norma ISO 14971, permitindo a avaliação contínua de perigos, modos de falha e eficácia das medidas de mitigação. Os dados provenientes de reclamações, eventos adversos, relatórios de tendências e acompanhamento clínico também apoiam a gestão de CAPA e a melhoria contínua.
04. Que papel desempenham as evidências do mundo real (RWE) na PMS?
As evidências do mundo real reforçam o PMS ao fornecer informações sobre a utilização efetiva dos dispositivos, incluindo o feedback dos doentes, dados de assistência técnica, registos e fontes de saúde digitais. As evidências do mundo real ajudam a identificar tendências, a validar o desempenho a longo prazo, a apoiar atualizações do perfil risco-benefício e a fundamentar decisões clínicas ou regulatórias. As entidades reguladoras esperam, cada vez mais, que os fabricantes incorporem as evidências do mundo real no PMS e na avaliação contínua pós-comercialização.
05. Quando é necessário realizar um PMCF para um dispositivo médico?
É necessário realizar PMCF quando as evidências clínicas existentes são insuficientes para confirmar a segurança ou o desempenho a longo prazo, ou quando a tecnologia, os materiais ou a utilização prevista de um dispositivo sugerem potenciais riscos a longo prazo. PMCF também desencadeado por novas tendências, riscos emergentes, incertezas clínicas, novos resultados da Avaliação de Riscos para a Saúde (HHE) ou quando as entidades reguladoras exigem a geração contínua de evidências para dispositivos de alto risco ou inovadores.
06. O que desencadeia um recall de um dispositivo médico ou uma ação corretiva no terreno (FCA)?
Uma recolha ou FCA é desencadeada quando um dispositivo representa um risco de segurança potencial ou confirmado, não cumpre os requisitos regulamentares ou apresenta problemas de desempenho que possam comprometer os resultados clínicos. Entre os fatores desencadeantes mais comuns contam-se tendências de defeitos, incidentes graves, erros de rotulagem, falhas de fabrico, vulnerabilidades de cibersegurança ou novas evidências que indiquem que o perfil benefício-risco do dispositivo se alterou.
07. Por que razão a Freyr é considerada um parceiro de referência em serviços de vigilância pós-comercialização?
A Freyr é amplamente reconhecida pela sua profunda experiência em matéria de regulamentação, cobertura do mercado global e abordagem estruturada à vigilância pós-comercialização (PMS), em conformidade com os requisitos EU MDR, FDA e da região Ásia-Pacífico. As organizações valorizam a Freyr pela sua combinação de conhecimento especializado, metodologias baseadas em dados, capacidades multilingues e um historial comprovado no apoio a diversos tipos de dispositivos, tornando-a um parceiro de confiança para operações de vigilância pós-comercialização fiáveis e em conformidade.







