
Visão geral dos Serviços de Conformidade com o IVDR da UE
O Regulamento Europeu de Dispositivos Médicos para Diagnóstico In Vitro (EU IVDR) é uma nova base regulamentar para a colocação de IVDs no mercado europeu. O Regulamento EU IVDR irá substituir a atual Diretiva da UE relativa aos dispositivos médicos para diagnóstico in vitro (UE 98/79/CE). Como regulamento europeu, será eficaz em todos os Member States da UE e nos estados da Associação Europeia de Comércio Livre (EFTA) sem necessidade de ser transposto para a legislação dos respetivos Estados. Como parceiro regulamentar comprovado, a Freyr fornece serviços de conformidade com o EU IVDR e serviços de consultoria IVDR a fabricantes para regulamentos de diagnóstico in vitro e marcação CE conformes.
Agende uma reunião com os nossos especialistas em IVDR da UE
O Regulamento IVDR - Cronograma de Transição
Com entrada em vigor obrigatória a partir de 26 de maio de 2022, o novo IVDR da UE é considerado uma alteração significativa à supervisão regulamentar dos IVD. A visão geral da transição do IVDR é a seguinte –
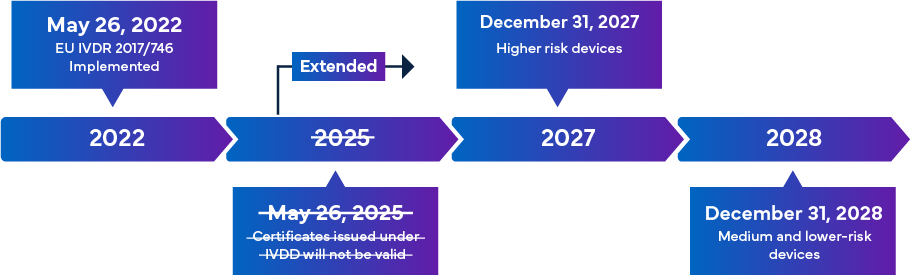
Uma vez totalmente implementadas, as regras de conformidade com o IVDR garantem que quase todos os IVD que entram no mercado europeu estão sujeitos à revisão do Organismo Notificado (ON) do IVDR e à certificação de marcação CE como parte do Processo de Aprovação de IVD. Neste cenário, para além de se alinharem com a classificação IVDR atualizada, existe uma exigência imediata para que os fabricantes revejam eficazmente a documentação técnica essencial para um registo e marcação CE de IVD bem-sucedidos. Os requisitos do IVDR variam com a classe de risco do IVD. Em geral, para a Certificação de IVD, os fabricantes devem realizar:
- Revisões do ficheiro técnico de acordo com o regulamento IVDR (Regulamento UE 2017/746)
- Preparação do Relatório de Avaliação de Desempenho (PER) para todos os IVD
- Acompanhamento do Desempenho Pós-Comercialização (PMPF) de acordo com o Anexo XIII, Parte B do IVDR
- Notificação de Vigilância de acordo com o Artigo 82 do IVDR
A Freyr apoia os clientes na realização de uma revisão sistemática da literatura científica e auxilia no planeamento e preparação de um PER para a conformidade com o Regulamento de Diagnóstico In Vitro. A Freyr tem especialistas dedicados que fornecem serviços de consultoria IVDR e suporte de Post-market Surveillance (PMS), que é uma parte integrante do sistema de gestão da qualidade do fabricante, juntamente com o Acompanhamento do Desempenho Pós-comercialização (PMPF).
Obtenha aconselhamento especializado sobre a sua conformidade com o IVDR da UE
Experiência e Vantagens dos Serviços de Conformidade com o IVDR da UE
- Plano de transição para a conformidade com o IVDR
- Revisão técnica e análise de lacunas dos requisitos do IVDR para os RGSPS (Requisitos Gerais de Segurança e Desempenho)
- Apoio na compilação do ficheiro técnico de acordo com os requisitos do IVDR
- Relatórios de validade científica com base na literatura e/ou dados internos
- Relatórios de desempenho clínico com base na literatura e/ou dados internos
- Relatórios de evidência clínica ou de avaliação de desempenho
- Protocolos e relatórios de acompanhamento do desempenho pós-comercialização (PMPF)
- Protocolos e relatórios de Post-market Surveillance (PMS).
- Elaboração/revisão de outros documentos, como folheto informativo/IFU (Instruções de Utilização), Instruções de Referência Rápida (QRI), Manual de Operação/Utilizador, etc.

- Garantia de conformidade com o IVDR, registo de IVD e marcação CE
- Sólido conhecimento e experiência em regulamentação nas principais áreas de impacto do Regulamento IVDR da UE
- Modelo de entrega impulsionado por uma forte gestão de projetos para garantir o cumprimento dos prazos
- Especialistas NB internos (revisão do relatório pelos revisores interativos do NB)
- Equipas focadas com experiência transversal em áreas de impacto específicas e categorias de dispositivos
- Contribuições multifuncionais de especialistas em dispositivos médicos para cumprir os requisitos
- Abrangência total de serviços em conformidade, revisão e planeamento
- Sólida experiência na manutenção da consistência nos resultados (Prazo e Qualidade)








