
Protocolo de pesquisa bibliográfica e visão geral da revisão sobre Diagnóstico In Vitro (IVD) e Dispositivos Médicos
No complexo panorama dos dispositivos médicos e dos dispositivos de diagnóstico in vitro (IVD), um protocolo de pesquisa bibliográfica bem estruturado para dispositivos médicos é mais do que um simples exercício de investigação; é um requisito fundamental para garantir a conformidade com EU MDR e com o Regulamento (UE) n.º 2017/746 relativo aos dispositivos de diagnóstico in vitro.
Uma revisão exaustiva da literatura médica apoia as avaliações clínicas e de desempenho, as atividades pós-comercialização e os pedidos de aprovação regulamentar. Permite a transparência, a rastreabilidade e a reprodutibilidade, que são essenciais tanto para cumprir as expectativas regulamentares como para a descoberta de evidências impulsionada pela IA — elementos-chave enfatizados pelos reguladores internacionais e essenciais para a descoberta de evidências através de pesquisas baseadas na IA
Requisitos de estado da técnica ao abrigo EU MDR do IVDR EU MDR
Tanto ao abrigo EU MDR do IVDR da UE, a determinação do estado da técnica (SOTA) constitui um requisito obrigatório para a avaliação clínica e de desempenho. O SOTA representa o nível atual e geralmente aceite de conhecimentos científicos, técnicos e clínicos relevantes para o dispositivo ou o IVD.
Um protocolo de pesquisa bibliográfica rigoroso é essencial para:
![]() Identificar tecnologias de referência e padrões de tratamento
Identificar tecnologias de referência e padrões de tratamento![]() Estabelecer perfis de segurança e desempenho aceites
Estabelecer perfis de segurança e desempenho aceites![]() Comparar o dispositivo em questão com as alternativas atuais
Comparar o dispositivo em questão com as alternativas atuais![]() Apoiar as atividades relacionadas com o CER, o PER, o CEP, o PEP, o PMS e PMCF
Apoiar as atividades relacionadas com o CER, o PER, o CEP, o PEP, o PMS e PMCF![]() Reunir provas sólidas para justificar a relação risco-benefício
Reunir provas sólidas para justificar a relação risco-benefício
 Identificar tecnologias de referência e padrões de tratamento
Identificar tecnologias de referência e padrões de tratamento Estabelecer perfis de segurança e desempenho aceites
Estabelecer perfis de segurança e desempenho aceites Comparar o dispositivo em questão com as alternativas atuais
Comparar o dispositivo em questão com as alternativas atuais Apoiar as atividades relacionadas com o CER, o PER, o CEP, o PEP, o PMS e PMCF
Apoiar as atividades relacionadas com o CER, o PER, o CEP, o PEP, o PMS e PMCF Reunir provas sólidas para justificar a relação risco-benefício
Reunir provas sólidas para justificar a relação risco-benefícioA Freyr garante que o seu conjunto de provas demonstre plenamente a conformidade com as expectativas do SOTA, um fator essencial para a aceitação por parte do Organismo Notificado.
Revisão EU MDR sobre o IVDR da UE e EU MDR
A revisão da literatura EU IVDR/ EU MDR é um componente crítico na gestão do ciclo de vida de um dispositivo médico ou IVD. Uma estratégia sistemática de pesquisa de literatura EU IVDR/ EU MDR fornece a base para os Clinical evaluation reports (CER), Relatórios de Avaliação de Desempenho (PER), Vigilância Pós-Comercialização (PMS), atividades de PMCF/PMPF ancoradas na literatura de IVD/dispositivos médicos baseada em evidências, este processo permite aos fabricantes apoiar a avaliação contínua da segurança e do desempenho.
A revisão EU MDR sobre o IVDR da UE EU MDR inclui normalmente:
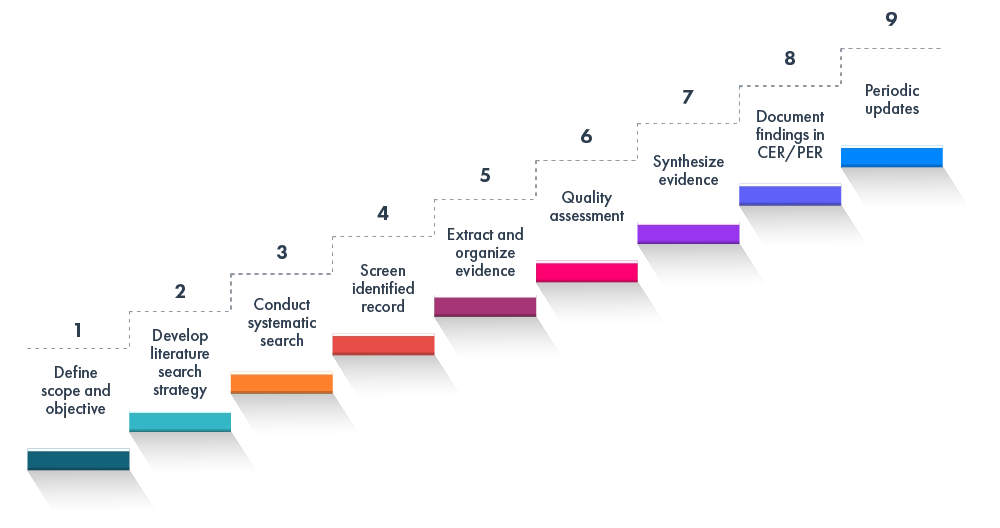
Este quadro está em conformidade com as melhores práticas globais em matéria de avaliações clínicas/de desempenho e revisões da literatura.
Principais diferenças entre os requisitos de pesquisa bibliográfica do IVDR e do MDR
Embora o MDR e o IVDR tenham em comum a avaliação sistemática das evidências, os seus requisitos diferem
O MDR (Dispositivos Médicos) centra-se em
![]() Avaliação clínica e evidência clínica
Avaliação clínica e evidência clínica![]() Declarações relativas à segurança e ao desempenho
Declarações relativas à segurança e ao desempenho![]() Recolha de PMCF
Recolha de PMCF![]() Justificação da relação benefício-risco
Justificação da relação benefício-risco![]() Alinhamento com a MEDDEV 2.7/1 Rev. 4
Alinhamento com a MEDDEV 2.7/1 Rev. 4
 Justificação da relação benefício-risco
Justificação da relação benefício-riscoO IVDR (Diagnóstico In Vitro) centra-se em
![]() Validade científica
Validade científica![]() Desempenho analítico
Desempenho analítico![]() Desempenho clínico
Desempenho clínico![]() Desenvolvimento do PER e do PEP
Desenvolvimento do PER e do PEP![]() Requisitos de comprovação do PMPF
Requisitos de comprovação do PMPF![]() Uma reclassificação mais rigorosa, que exige mais provas de apoio
Uma reclassificação mais rigorosa, que exige mais provas de apoio
 Validade científica
Validade científica Desempenho analítico
Desempenho analítico Desenvolvimento do PER e do PEP
Desenvolvimento do PER e do PEP Requisitos de comprovação do PMPF
Requisitos de comprovação do PMPF Uma reclassificação mais rigorosa, que exige mais provas de apoio
Uma reclassificação mais rigorosa, que exige mais provas de apoioA Freyr adapta as estratégias de pesquisa bibliográfica, o desenvolvimento de CER/PER e os protocolosPMCF de acordo com o processo regulamentar do dispositivo.
O Poder de uma Equipa Robusta de Síntese de Literatura Científica
Cumprir os requisitos do MDR e do IVDR exige mais do que simples pesquisas em bases de dados. Uma equipa especializada em síntese de literatura científica, com conhecimentos terapêuticos, garante que a sua revisão de literatura relativa ao IVDR/MDR, o protocolo de pesquisa bibliográfica e a documentação de avaliação clínica/de desempenho cumprem os padrões de profundidade e rigor exigidos pelas entidades reguladoras.
Os especialistas da Freyr simplificam vias complexas e transformam dados clínicos, de desempenho e científicos em evidências claras e fundamentadas que reforçam os protocolos de pesquisa bibliográfica, as avaliações comparativas de eficácia (CER), as avaliações comparativas de resultados (PER) e as estratégias de gestão de seguros de saúde (PMS/PMPF).
Através de metodologias sistemáticas, técnicas de pesquisa avançadas e competências de avaliação crítica, a nossa equipa garante que todas as revisões da literatura cumprem as expectativas regulamentares globais, ao mesmo tempo que melhora a qualidade, a credibilidade e a prontidão do seu conjunto de evidências, conferindo ao seu dispositivo uma forte vantagem competitiva num mercado em rápida evolução.
Protocolo de pesquisa EU MDR sobre o IVDR da UE / EU MDR
Um protocolo de pesquisa EU MDR em conformidade com o IVDR e EU MDR proporciona uma estrutura, reduz o enviesamento dos revisores e garante a total rastreabilidade durante as auditorias.
O processo do protocolo de pesquisa bibliográfica sobre o IVDR/MDR inclui:
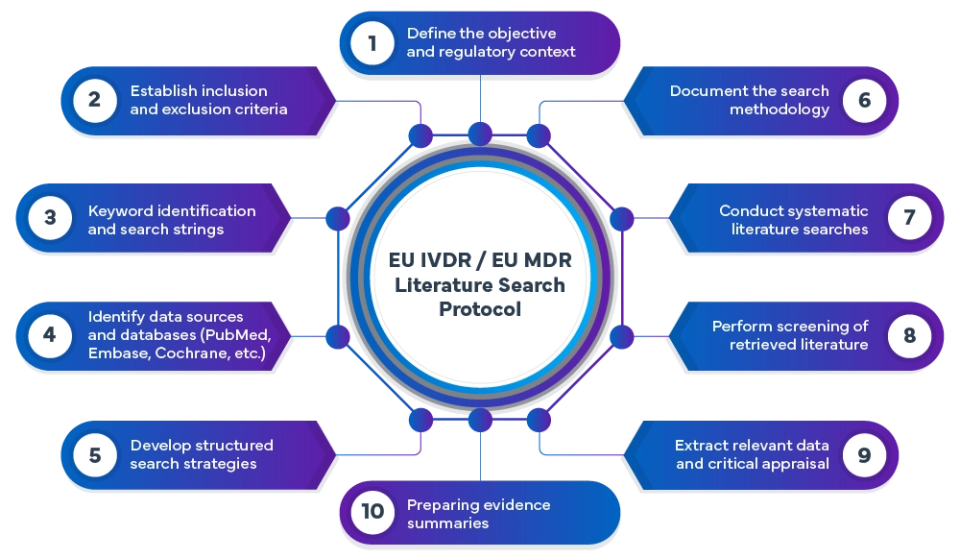
Na Freyr, realizamos análises bibliográficas exaustivas, em conformidade com o MDR/IVDR, utilizando metodologias de pesquisa avançadas. As publicações presentes em bases de dados globais são sistematicamente selecionadas e analisadas para identificar evidências relevantes que comprovem a segurança, o desempenho e os benefícios clínicos dos dispositivos.
Protocolo e revisão da pesquisa bibliográfica sobre diagnósticos in vitro (IVD) e dispositivos médicos
- Identificação, compilação e síntese sistemáticas da literatura científica
- Conceção e implementação de protocolos de pesquisa bibliográfica em conformidade com o MDR/IVDR
- Definição de questões de investigação e estratégias de pesquisa personalizadas para o dispositivo.
- Identificação de palavras-chave, criação de cadeias de pesquisa e seleção de bases de dados (PubMed, Embase, Cochrane, etc.)
- Avaliação crítica das evidências clínicas, de desempenho e científicas.
- Resumo de evidências para documentação regulamentar.
- Elaboração de CERs, CEPs, PERs e PEPs
- Avaliação das lacunas nos dados da documentação existente relativa a CER/CEP/PEP/PER.
- Utilizando técnicas de pesquisa avançadas para identificar a literatura global relevante

- Conformidade garantida com o MDR/IVDR
- Processo de pesquisa bibliográfica estruturado, reproduzível e fundamentado
- Estratégias de evidência personalizadas e específicas para cada dispositivo
- Especialistas clínicos e regulamentares altamente qualificados
- Capacidade escalável da equipa Estratégias de evidência personalizadas e específicas para cada dispositivo
- Contribuições interdisciplinares nas áreas regulamentar, médica e clínica
- Pesquisa End-to-end , revisão e apoio à documentação
- Aumenta a credibilidade, a clareza e a prontidão dos pedidos de aprovação regulamentar

Perguntas Frequentes (FAQs)
01. Qual é o objetivo de um protocolo de pesquisa bibliográfica sobre dispositivos médicos ao abrigo EU MDR?
Um protocolo de pesquisa bibliográfica sobre dispositivos médicos proporciona uma abordagem estruturada, sistemática e transparente para identificar, avaliar e documentar as evidências científicas relativas a um dispositivo ou aos seus comparadores. Garante a reprodutibilidade, minimiza o enviesamento e permite que as entidades reguladoras acompanhem a forma como as evidências clínicas ou de desempenho foram recolhidas, avaliadas e sintetizadas. Ao abrigo EU MDR, esse protocolo apoia as avaliações de segurança, desempenho e relação benefício-risco, garante a conformidade regulamentar e constitui a base para uma documentação de alta qualidade e defensável ao longo de todo o ciclo de vida do dispositivo.
02. De que forma os avanços mais recentes influenciam a avaliação clínica e de desempenho?
O «estado da arte» (SOTA) representa o conhecimento científico e clínico atualmente aceite para um determinado tipo de dispositivo. Estabelece uma referência em termos de segurança, desempenho e resultados clínicos esperados. A definição do SOTA através da revisão da literatura ajuda a contextualizar as alegações relativas ao dispositivo, apoia a seleção de comparadores e orienta a avaliação da relação benefício-risco, o planeamento PMCF e as atualizações das evidências ao longo do ciclo de vida.
03. O que distingue uma revisão da literatura MDR de uma revisão sistemática tradicional?
Uma revisão bibliográfica MDR/IVDR difere de uma revisão sistemática tradicional, na medida em que se centra nos requisitos regulamentares e é especificamente concebida para apoiar a conformidade com esses requisitos. Enquanto as revisões sistemáticas tradicionais visam responder a questões de investigação científica e servem fins puramente académicos, uma revisão bibliográfica MDR avalia evidências clínicas e de desempenho para demonstrar a segurança, o desempenho e os perfis de risco-benefício dos dispositivos. Segue uma metodologia estruturada e rastreável, com questões de investigação predefinidas, critérios de inclusão/exclusão e avaliação crítica, para produzir documentação defensável e pronta para auditoria para submissões regulamentares
04. Com que frequência devem ser atualizadas as revisões da literatura relativas a dispositivos médicos e dispositivos de diagnóstico in vitro?
A frequência das atualizações depende do risco do dispositivo, da dinâmica do mercado e da evolução dos dados disponíveis. Os dispositivos de alto risco requerem normalmente atualizações anuais, enquanto outros podem seguir intervalos definidos. As revisões devem também ser atualizadas sempre que surjam sinais de segurança significativos, novos dados clínicos, avanços tecnológicos ou alterações nas diretrizes, a fim de manter perfis de risco-benefício precisos.
05. Que papel desempenham os critérios de inclusão e exclusão nas pesquisas bibliográficas sobre a MDR/IVDR?
Os critérios de inclusão e exclusão garantem que apenas sejam selecionadas evidências relevantes e de alta qualidade. Melhoram a objetividade, reduzem o viés dos revisores e asseguram uma tomada de decisão consistente. Ao abrigo do MDR/IVDR, estes critérios devem ser predefinidos, justificados e alinhados com as questões de investigação, a fim de manter a rastreabilidade e a defensabilidade regulamentar ao longo de todo o processo de avaliação.
06. Por que razão a avaliação crítica é essencial nas revisões da literatura sobre o MDR/IVDR?
A avaliação crítica analisa a qualidade metodológica, a relevância e a fiabilidade das evidências incluídas. Os quadros regulamentares da MDR/IVDR dão ênfase à avaliação, uma vez que as entidades reguladoras se baseiam em alegações de segurança e desempenho devidamente fundamentadas. Uma avaliação rigorosa ajuda a distinguir dados sólidos de estudos menos robustos e reforça as conclusões utilizadas nas CER, PER, relatórios de PMS e análises de risco-benefício.
07. Em que diferem os requisitos de pesquisa bibliográfica do MDR e do IVDR?
O MDR centra-se na avaliação clínica, na justificação da relação benefício-risco e no desempenho clínico, enquanto o IVDR dá ênfase ao desempenho analítico, à validade científica e ao desempenho clínico no que diz respeito à precisão do diagnóstico. As estratégias de revisão da literatura devem refletir estas diferenças, adaptando as questões de investigação, os conjuntos de dados e os quadros de avaliação às distintas vias de evidência exigidas por cada regulamento.
08. Que bases de dados e fontes de informação devem ser consideradas nas pesquisas bibliográficas alinhadas com o MDR/IVDR?
As entidades reguladoras esperam que sejam utilizadas várias bases de dados científicas, tais como PubMed, Embase e Cochrane, complementadas por bases de dados de vigilância, registos de ensaios clínicos, diretrizes e literatura cinzenta relevante. O recurso a fontes diversificadas garante uma cobertura abrangente das informações clínicas, de desempenho e de segurança necessárias para uma avaliação sólida e uma vigilância contínua.
09. Por que razão a Freyr é considerada um parceiro de referência na pesquisa bibliográfica e na elaboração de protocolos?
A Freyr é considerada um parceiro de referência devido ao seu profundo conhecimento da regulamentação, rigor científico e cumprimento consistente das exigências de evidência do MDR/IVDR. A equipa aplica princípios de revisão sistemática, uma metodologia transparente e conhecimentos especializados na área terapêutica para produzir resultados defensáveis e prontos para auditoria. A abordagem da Freyr privilegia a rastreabilidade, a avaliação crítica e a comparação com os padrões de excelência mais recentes, fatores-chave valorizados pelos organismos notificados e pelas entidades reguladoras globais.







