3 Min. Lesezeit
Die Kennzeichnung ist ein integraler Bestandteil der Vermarktung von Medizinprodukten. Das Etikett ist eine Information, die am Gerät und/oder an der Verpackung in einem für Menschen lesbaren Format angebracht ist. Der Hauptzweck der Kennzeichnung besteht darin, Sicherheitsinformationen für Benutzer bereitzustellen, bei denen es sich um medizinisches Fachpersonal, Verbraucher oder andere relevante Personen handeln kann.
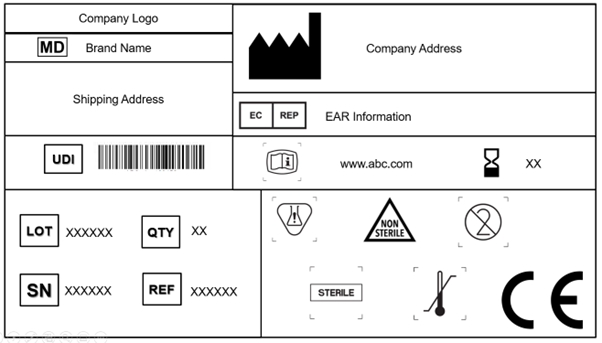
Alle globalen Regulierungsbehörden haben bestimmte Kennzeichnungsanforderungen. Ebenso hat die EU die Kennzeichnungsanforderungen in Kapitel III unter Anhang I der EU-Medizinprodukte-Verordnung (EU MDR) 2017/745 detailliert beschrieben. Das Wichtigste ist, alle Symbole, die die erforderlichen Informationen abdecken, in die Kennzeichnung des Geräts und die begleitenden Dokumente (Broschüren, Handbücher, Gebrauchsanweisungen usw.) aufzunehmen.
Einige der kritischen Überlegungen zur Kennzeichnung, die bei der Einhaltung der EU MDR 2017/745 zu beachten sind, sind-
1. Kennzeichnungssymbolik für Medizinprodukte
Jeder Hersteller ist verpflichtet, das Symbol für Medizinprodukte anzubringen, das besagt, dass das auf dem EU-Markt bereitgestellte Produkt ein Medizinprodukt ist. Es ist zwingend erforderlich, dieses Symbol auf dem Produkt und allen Verpackungsebenen anzubringen. Zusätzlich sollte das Etikett den Handelsnamen und den ursprünglichen Namen des Produkts angeben.
2. Spezielle Geräte
Handelt es sich um eine Sonderanfertigung oder ein kundenspezifisches Produkt, sollte dessen Status auf der Kennzeichnung angegeben werden. Wenn das Produkt beispielsweise nur für klinische Prüfungen bestimmt ist, sollte dies auf dem Etikett ausdrücklich vermerkt sein.
Bei Produkten mit absorbierenden Materialien oder solchen, die sich lokal im menschlichen Körper verteilen können, sollte die Kennzeichnung die Zusammensetzung des Materials sowie quantitative Angaben zu den Hauptbestandteilen enthalten.
Auch bei Einmalprodukten und sterilen Produkten ist eine explizite Kennzeichnung erforderlich. Bei wiederaufbereiteten Produkten sollte die Kennzeichnung die maximale Anzahl der Wiederaufbereitungen, die bisherige Anzahl der Wiederaufbereitungen und die verwendete Sterilisationsmethode angeben.
3. Vorhandensein toxischer Substanzen
Die Angabe des Vorhandenseins von CMR-Stoffen (karzinogen, mutagen, reproduktionstoxisch) und endokrin wirksamen Substanzen ist auf den Etiketten zwingend vorgeschrieben, wenn die Konzentration mehr als 0,1 % (Gew./Gew.) beträgt. Die Liste dieser Substanzen muss am Produkt und/oder an der Verpackung angebracht werden.
Des Weiteren muss ein Etikett über das Vorhandensein von Blut- und Gewebederivaten (auch wenn diese in der medizinischen Substanz des Kombinationsprodukts enthalten sind) an den Produkten angebracht werden.
4. Harmonisierte Normen
Die EU MDR 2017/745 erkennt die ISO 15223-1: 2021 an und akzeptiert sie. Das Dokument legt die Symbole fest, die bei der Kennzeichnung von Medizinprodukten und deren Verpackung zu verwenden sind. Kapitel 3 (23.1,h) von Anhang I der EU MDR legt fest, dass international anerkannte Symbole verwendet werden können, und in Regionen, in denen diese Symbole nicht anerkannt sind, muss eine Beschreibung derselben in einem Dokument zusammen mit dem Gerät bereitgestellt werden.
5. UDI
Die Artikel 27, 28, 29 und Anhang VI (A, B, C) legen die Regeln und Vorschriften für die UDI detailliert fest. Das Etikett muss nun einen UDI-Träger [Darstellung der UDI zur automatischen Identifizierung und Datenerfassung (AIDC) und zur menschenlesbaren Interpretation (HRI)] auf dem Produkt und auch auf höheren Verpackungsebenen enthalten. Die höhere Verpackung des Produkts (ausgenommen Versandverpackungen) wird einen eigenen UDI-Träger aufweisen.
6. Elektronische Gebrauchsanweisung (eIFU)
Webadressen (URLs) in Form von eIFUs können neben den gedruckten Gebrauchsanweisungen (IFUs) auch auf der Kennzeichnung von Medizinprodukten angebracht werden. Die eIFUs können bei implantierbaren, aktiven implantierbaren, festen Medizinprodukten und Software (auch für Laien bestimmt) verwendet werden.
7. Informationen der Wirtschaftsakteure (EOs)
Das Etikett enthält in der Regel die Informationen des Herstellers. Bei ausländischen Herstellern sollten jedoch die Informationen des Bevollmächtigten auf den kommerziellen Etiketten angebracht werden.
8. Warnhinweise und Vorsichtsmaßnahmen
Die Warnhinweise und Vorsichtsmaßnahmen müssen auf dem Etikett des Produkts angegeben werden. Die Informationen hierzu können auf ein Minimum beschränkt werden, und Details können in der Gebrauchsanweisung (IFU) bereitgestellt werden.
Die Hersteller müssen sich auch an die länderspezifischen Kennzeichnungsanforderungen anpassen. Die Sprachanforderung hängt vom EU-Mitgliedstaat ab. Dies kann die Kennzeichnung, die Gebrauchsanweisungen (IFUs) und die Verpackung des Produkts erheblich in Bezug auf Zeit und Kosten beeinflussen.
Diese zusätzlichen Anforderungen können die Belastung des Herstellers angesichts der bestehenden Komplexität des Kennzeichnungsprozesses weiter erhöhen. Ein Versäumnis in diesem Bereich kann sehr kostspielig werden und Produktrückrufe sowie nachfolgende Schritte für Korrektur- und Vorbeugemaßnahmen (CAPA) nach sich ziehen.
Suchen Sie Unterstützung bei der Kennzeichnung gemäß der EU MDR? Freyr bietet umfassende Dienstleistungen für die Kennzeichnung von Medizinprodukten. Kontaktieren Sie unsere Regulierungsexperten jetzt unter – sales@freyrsolutions.com









