
5 min czytania
Program Jednolitego Audytu Wyrobów Medycznych (MDSAP) umożliwia uznanej Organizacji Audytowej (AO) przeprowadzenie pojedynczego audytu Systemu Zarządzania Jakością (QMS) producenta wyrobów medycznych. Dostarcza on odpowiednie wymogi regulacyjne dla pięciu krajów, tj. Brazylii (ANVISA), USA (FDA), Japonii (PMDA), Kanady (Health Canada) i Australii (TGA). Oprócz uczestniczących organów regulacyjnych, w MDSAP zaangażowanych jest kilku innych międzynarodowych partnerów (oficjalni obserwatorzy i członkowie stowarzyszeni).
Certyfikacja MDSAP jest obowiązkowa w Kanadzie (Health Canada) dla wyrobów klasy II, III i IV, ale dobrowolna dla pozostałych czterech krajów. Promuje ona przejrzystość i zgodność regulacyjną między uczestniczącymi organami oraz minimalizuje potrzebę przeprowadzania wielu audytów, oszczędzając w ten sposób czas i zasoby producentów wyrobów medycznych. Aby lepiej przedstawić program MDSAP, zebraliśmy tutaj odpowiedzi na piętnaście (15) najczęściej zadawanych pytań.
- Dlaczego Program MDSAP został opracowany, skoro istnieje globalnie akceptowana certyfikacja ISO 13485?
MDSAP został opracowany w celu zmniejszenia obciążenia związanego z audytami regulacyjnymi dla producentów wyrobów medycznych oraz w celu promowania większej spójności podejść regulacyjnych i wymagań technicznych opartych na międzynarodowych standardach i najlepszych praktykach. Koncentruje się na zapewnieniu spójności, przewidywalności i przejrzystości programów regulacyjnych poprzez standaryzację procedur i praktyk organów regulacyjnych oraz zewnętrznych organizacji audytujących.
Audyt opiera się na wymaganiach QMS zgodnie z ISO 13485 oraz wymaganiach regulacyjnych kraju uczestniczącego, w którym wyroby medyczne będą wprowadzane do obrotu.
- Jakie są kryteria kwalifikacji do poddania się audytowi MDSAP?
Każdy producent wyrobów medycznych zamierzający wprowadzić swój wyrób na rynki krajów uczestniczących, może poddać się audytowi MDSAP. Jednakże, każda jednostka regulacyjna może w razie potrzeby ustanowić kryteria wykluczenia dla określonych warunków.
Na przykład w Japonii wyjątki od kryteriów kwalifikowalności to:
- Zarejestrowany Zakład Produkcyjny (RMS), który wytwarza wyroby medyczne wykonane z tkanek ludzkich/zwierzęcych
- RMS, który wytwarza radioaktywne IVD, oraz
- Ustanowienie Posiadacza Pozwolenia na dopuszczenie do obrotu produktu leczniczego (MAH)
- Czy audyt MDSAP obejmuje produkty złożone?
Wyroby medyczne, które zawierają leki (substancje lecznicze) lub produkty biologiczne (np. materiały pochodzenia zwierzęcego, które zostały unieszkodliwione, lub tkanki, komórki, lub substancje pochodzenia mikrobiologicznego lub rekombinowanego, ludzka krew lub ekstrakty ludzkiej krwi lub produkty krwiopochodne itp.) są uważane za produkty złożone i mogą być objęte zakresem audytu MDSAP.
Jednakże, ze względu na różnice w sposobie regulowania tych produktów w jurysdykcjach uczestniczących organów regulacyjnych, raporty z audytów MDSAP i dokumenty certyfikacyjne mogą nie być uznawane za alternatywę dla wymagań inspekcyjnych i oceny w niektórych jurysdykcjach.
Australia- Produkty złożone podlegają zdalnej ocenie TGA w ramach Australijskiej Oceny Zgodności. Skuteczny audyt MDSAP może jednak zmniejszyć liczbę inspekcji dla tych urządzeń.
Brazylia, Japonia- Produkty złożone uznawane za wyroby medyczne są objęte MDSAP, ponieważ nie ma specyficznych wymagań dotyczących QMS.
Kanada- Model MDSAP obejmuje wymagania QMS dla produktów złożonych uznawanych za wyroby medyczne.
USA- Audyty MDSAP nie są uważane za alternatywę dla inspekcji FDA w przypadku produktów złożonych.
- Czy mogę wybrać kraj objęty audytem MDSAP?
Tak, audyt jest przeprowadzany zgodnie z zakresem zadeklarowanym we wniosku o usługi certyfikacyjne. Od producentów wyrobów medycznych oczekuje się zgodności z przepisami wyłącznie w jurysdykcjach, w których ich produkty mają być wprowadzane do obrotu.
- Jestem producentem wyrobów medycznych ze US, zamierzającym wprowadzić mój wyrób na rynek wyłącznie w Japonii. Wkrótce przejdę audyt MDSAP. Czy muszę również spełniać wymagania innych krajów?
Nie, od producentów wyrobów medycznych oczekuje się jedynie zgodności z wymaganiami i przepisami ISO 13485 w jurysdykcjach, w których ich produkty mają być wprowadzane na rynek.
- Moja Organizacja Audytująca (AO) i Europejska Jednostka Notyfikowana są takie same. Czy mogę być audytowany w zakresie obu jednocześnie?
Jeśli Twoja AO i Europejska Jednostka Notyfikowana są takie same, ocena zgodności może być przeprowadzona po audycie MDSAP, a nie jednocześnie. Europejskie Jednostki Notyfikowane są obserwatorami MDSAP, a ocena zgodności jest przeprowadzana zgodnie z EU MDR 2017/745. W przypadku MDSAP, ocena jest przeprowadzana zgodnie z wymaganiami ISO 13485 i wymaganiami regulacyjnymi uczestniczących krajów objętych zakresem.
- Jaka jest różnica między ocenami Etapu I i II?
Początkowy proces audytu MDSAP obejmuje dwa etapy. Audyt początkowy, zwany również audytem Certyfikacji Początkowej, składa się z audytów Etapu I i Etapu II.
Audyt Etapu I obejmuje przegląd dokumentacji oraz ocenę gotowości producenta wyrobu medycznego do poddania się audytowi Etapu II.
Audyt etapu II jest przeprowadzany w celu weryfikacji, czy wszystkie mające zastosowanie wymagania ISO 13485 oraz inne wymagania regulacyjne organu regulacyjnego objętego zakresem są wdrożone.
- Ilu audytorów mogę się spodziewać podczas audytu MDSAP?
Określenie czasu audytu precyzuje, jak ustalić czas trwania audytu na miejscu w osobo-dniach. AO decyduje ilu audytorów będzie tworzyć zespół audytowy. Na przykład, audyt trwający sześć (06) osobo-dni może zostać zakończony w trzy (03) dni przez zespół dwóch (02) audytorów.
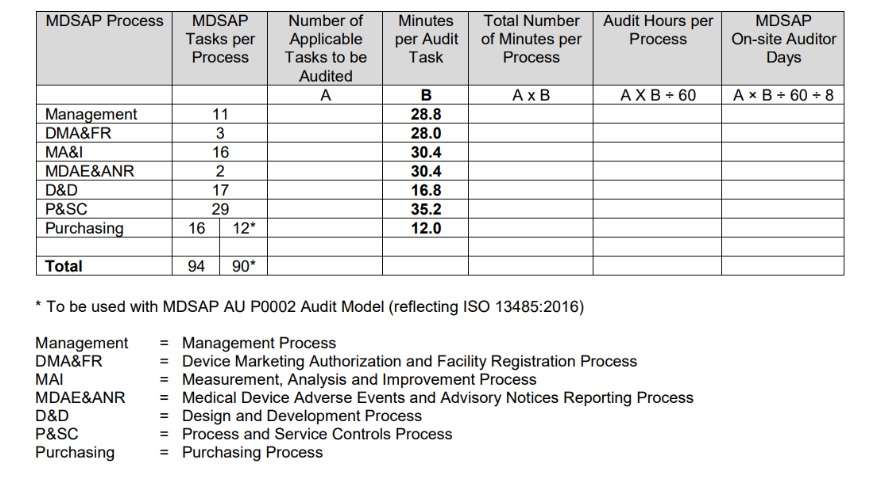
- Jak zaplanowany jest audyt MDSAP?
„Procedura Ustalania Czasu Audytu”, wydana przez FDA, podsumowuje proces ustalania czasu trwania audytu w poniższej tabeli.
Obliczenie czasu trwania audytu opiera się przede wszystkim na liczbie odpowiednich zadań audytowych związanych z rodzajem przeprowadzanej kontroli oraz konkretnymi działaniami organizacji podlegającymi audytowi.
W celu uzyskania szczegółowych informacji na ten temat, można zapoznać się z MDSAP P0008007
- Czy istnieje przewodnik lub lista kontrolna, do której mogę uzyskać dostęp, aby zapewnić zgodność z audytem MDSAP?
Tak, możesz uzyskać dostęp do dokumentu MDSAP Audit Approach. Jest to dobrze zorganizowany przewodnik wydany przez USFDA, który odwołuje się do konkretnych sekcji normy ISO 13485:2016 oraz odpowiednich przepisów wydanych przez australijską TGA, brazylijską ANVISA, kanadyjską Health Canada, japońską MHLW/PMDA i amerykańską FDA.
- Jaka jest rola obserwatora w audycie MDSAP?
Obserwator MDSAP to organ regulacyjny, który ma prawo uczestniczyć w spotkaniach, ocenach i innych działaniach, ale nie korzysta z rezultatów MDSAP. Obserwatorzy są reprezentowani w Radzie Organów Regulacyjnych MDSAP (RAC) przez jednego menedżera wyższego szczebla.
- Jakie są kolejne kroki, które należy podjąć, jeśli otrzymałeś ocenę 4 lub wyższą?
System ocen jest stosowany do niezgodności stwierdzonych podczas audytu przez AO. Ocena 4 lub 5 wskazuje na wysokie ryzyko interwencji. Należy przedstawić plan naprawczy dla każdej zarejestrowanej niezgodności w ciągu 15 dni kalendarzowych od daty wystawienia raportu o niezgodności. Plan naprawczy musi zawierać wyniki dochodzenia w sprawie niezgodności, jej przyczyny oraz zaplanowane działania korygujące, aby zapobiec jej ponownemu wystąpieniu. Dowody wdrożenia planu/działania naprawczego należy dostarczyć w ciągu trzydziestu (30) dni kalendarzowych od daty zakończenia audytu.
- Czy istnieje różnica w podejściu do audytu przez audytora wewnętrznego w porównaniu z AO?
MDSAP opiera się na podejściu procesowym. AO prawdopodobnie będzie analizować powiązania i zależności, podczas gdy audytor wewnętrzny może skupiać się na jednym aspekcie funkcjonalnym w danym momencie. Dlatego AO może znaleźć niezgodność w jednym obszarze funkcjonalnym i szukać odpowiedzi w innym obszarze funkcjonalnym. Jednakże stosowanie podejścia procesowego może być zakłócające podczas audytu wewnętrznego.
- Czy mogę odwołać się do AO, jeśli udowodnię, że zarejestrowana niezgodność jest nieważna?
AO posiada proces odwoławczy lub rozstrzygania sporów, który można wykorzystać, jeśli można wykazać, że zarejestrowana niezgodność jest nieważna. Jednakże, oceny przypisane niezgodnościom nie mogą być zmienione z powodu działań korygujących. Mogą być zmienione jedynie na podstawie dowodów wskazujących, że były one nieważne.
- Jak długo ważny jest certyfikat MDSAP?
Producenci wyrobów medycznych certyfikowani w ramach programu MDSAP będą poddawani corocznym audytom, zgodnie z trzyletnim cyklem certyfikacji. Audyt początkowy to pełny audyt systemu zarządzania jakością (QMS) producenta wyrobów medycznych. Następnie przeprowadzane są audyty nadzoru, realizowane corocznie przez dwa (02) kolejne lata. Cykl rozpoczyna się ponownie audytem recertyfikacyjnym w trzecim roku.
Aby dowiedzieć się więcej o naszych usługach MDSAP, skontaktuj się z Freyr już dziś, aby umówić rozmowę z naszymi Ekspertami.









