
3 minuty czytania
W nieustannie zmieniającej się i ściśle regulowanej branży produkcji wyrobów medycznych zapewnienie jakości produktów i bezpieczeństwa pacjentów to nie tylko obowiązek prawny, ale także strategiczna konieczność. Skuteczne systemy zarządzania reklamacjami i nadzoru, zgodne z ISO 13485:2016, stanowią podstawę każdego solidnego systemu zarządzania jakością (QMS). Mechanizmy te gwarantują zgodność z międzynarodowymi przepisami oraz wzmacniają zaufanie klientów i wiarygodność marki.
Przyjrzyjmy się, jak rozpatrywanie skarg i czuwanie nad bezpieczeństwem odgrywają kluczową rolę w utrzymaniu jakości i zgodności z przepisami w branży wyrobów medycznych, w oparciu o wytyczne zgodne z ISO 13485:2016 i poparte globalnymi ramami regulacyjnymi.
Zrozumienie zasad zarządzania reklamacjami zgodnie z ISO 13485:2016
ISO 13485:2016 definiuje skargę jako każdą formę komunikacji – pisemną, ustną lub elektroniczną – zawierającą zarzuty dotyczące tożsamości, jakości, trwałości, niezawodności, użyteczności, bezpieczeństwa lub działania wyrobu medycznego. Norma ta nakłada na producentów wyrobów medycznych obowiązek ustanowienia udokumentowanych procedur rozpatrywania skarg, obejmujących ocenę, badanie i rozstrzyganie skarg w ramach skutecznego systemu zarządzania jakością.
System obsługi reklamacji ma kluczowe znaczenie dla zapewnienia zgodności z przepisami, utrzymania zadowolenia klientów oraz poprawy jakości urządzeń. Proces ten musi być uporządkowany i przejrzysty oraz obejmować następujące etapy:
- Rejestracja i dokumentacja zgłoszeń
Wszystkie zgłoszenia należy rejestrować bezzwłocznie, podając jak najwięcej szczegółów dotyczących charakteru problemu, specyfikacji i modelu urządzenia, warunków jego użytkowania oraz wszelkich skutków odczuwanych przez użytkownika lub pacjenta. - Ocena pod kątem obowiązku zgłoszenia
Po zarejestrowaniu skarg należy je poddać ocenie w celu ustalenia, czy stanowią one zdarzenia podlegające zgłoszeniu. Na przykład w Stanach Zjednoczonych producenci wyrobów medycznych mają obowiązek zgłaszania określonych problemów związanych z wyrobami w ramach systemu zgłaszania wyrobów medycznych (MDR) regulowanego przez FDA. Producenci muszą przestrzegać obowiązujących przepisów dotyczących terminów zgłoszeń. - Dochodzenie i analiza przyczyn źródłowych
W przypadku uzasadnionych skarg należy przeprowadzić szczegółowe dochodzenie. Ustalenie przyczyny źródłowej ma zasadnicze znaczenie dla wdrożenia skutecznych działań korygujących i zapobiegawczych (CAPA). - Działania korygujące i zapobiegawcze (CAPA)
Na podstawie ustaleń dotyczących wyrobów medycznych przedsiębiorstwa muszą podjąć działania CAPA, aby rozwiązać zarówno bieżący problem, jak i zapobiec jego ponownemu wystąpieniu oraz uniknąć podobnych sytuacji w przyszłości. - Pętla sprzężenia zwrotnego i zamknięcie sprawy
Po rozwiązaniu problemu skarga powinna zostać formalnie zamknięta wraz z kompletną dokumentacją oraz powiadomieniem skarżącego, o ile ma to zastosowanie w przypadku wyrobu medycznego.
Czujność: podstawowy filar nadzoru po wprowadzeniu do obrotu
Nadzór oznacza monitorowanie i zgłaszanie zdarzeń niepożądanych oraz incydentów po wprowadzeniu wyrobu medycznego do obrotu. Zgodnie z ISO 13485:2016 i odpowiednimi przepisami międzynarodowymi ten ciągły nadzór ma zasadnicze znaczenie dla wczesnego wykrywania potencjalnych zagrożeń i podejmowania odpowiednich działań w odpowiednim czasie.
System nadzoru Unii Europejskiej, zaktualizowany na mocy rozporządzenia w sprawie wyrobów medycznych (EU MDR), nakłada na producentów wyrobów medycznych obowiązek posiadania planów nadzoru po wprowadzeniu do obrotu. Plany te muszą obejmować procedury gromadzenia i analizowania danych dotyczących rzeczywistego użytkowania wyrobu.
Podobnie w Indiach Central Drugs Standard Control Organization CDSCO) reguluje procedury nadzoru i rozpatrywania skarg dotyczących wyroby medyczne. Zachęca ona do zgłaszania zdarzeń niepożądanych i awarii produktów w celu ciągłego zwiększania bezpieczeństwa i skuteczności wyrobów medycznych.
Do kluczowych elementów skutecznego systemu nadzoru należą:
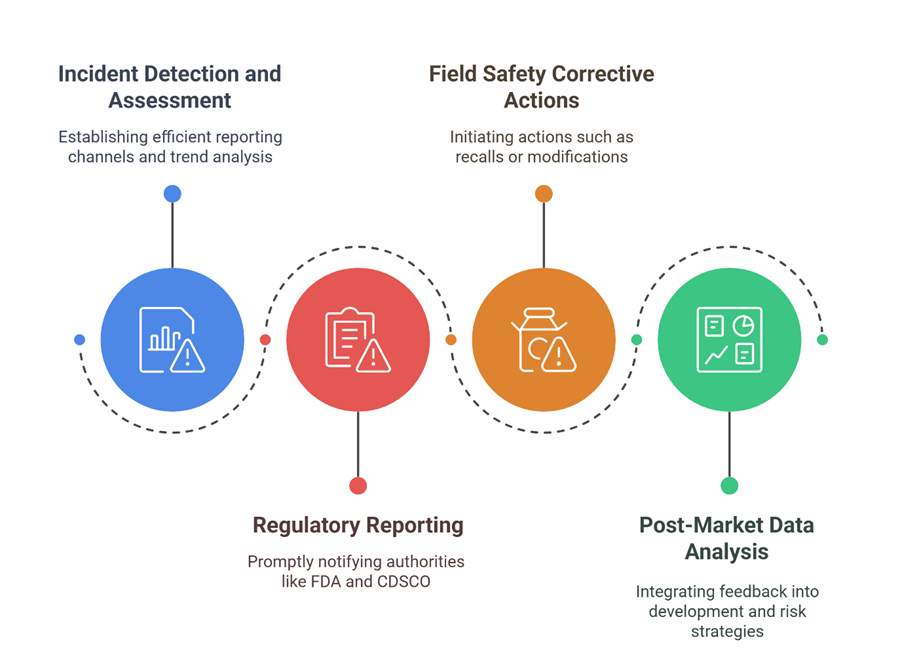
Opracowanie zintegrowanej strategii w zakresie rozpatrywania skarg i nadzoru
Producenci wyrobów medycznych mogą usprawnić swoje systemy zarządzania jakością poprzez włączenie obsługi reklamacji i nadzoru nad bezpieczewnością produktów do jednego spójnego systemu. Oto najlepsze praktyki:
Jasne procedury operacyjne i określenie zakresu obowiązków
Standardowe procedury operacyjne (SOP) powinny określać kolejne etapy przyjmowania, oceny, zgłaszania i rozpatrywania skarg. Określają one, kto jest odpowiedzialny na każdym etapie, aby uniknąć opóźnień i luk.
- Szkolenia i podnoszenie świadomości pracowników
Regularne szkolenia pracowników gwarantują, że personel wie, jak skutecznie rozpoznawać skargi i zdarzenia niepożądane oraz jak sobie z nimi radzić. - Narzędzia cyfrowe i automatyzacja
Wykorzystanie oprogramowania do zarządzania jakością (QMS) pozwala usprawnić śledzenie reklamacji, dokumentację i sprawozdawczość. Automatyzacja poprawia identyfikowalność i zgodność z ISO 13485:2016. - Kultura jakości i ciągłe doskonalenie
Propagowanie filozofii, w której ceni się informacje zwrotne, zachęca do aktywnego zgłaszania problemów i sprzyja innowacyjności w zakresie poprawy jakości.
Zgodność z przepisami i nie tylko: przewaga marketingowa
Wdrożenie systemów zarządzania skargami i nadzorem zgodnych z ISO 13485:2016 nie tylko gwarantuje przestrzeganie przepisów, ale także buduje wizerunek firmy jako podmiotu godnego zaufania i dbającego o jakość. W branży, w której bezpieczeństwo i wydajność mają bezpośredni wpływ na życie ludzi, takie zaangażowanie może stanowić potężne narzędzie marketingowe.
Zgodnie z FDA dostosowanie się do międzynarodowych standardów poprawia dostęp do rynków światowych, wzmacnia zaufanie klientów oraz zmniejsza ryzyko kosztownych wycofań produktów z rynku lub sporów sądowych.
| Region | Ramy regulacyjne | Numer klauzuli odniesienia / artykuł dotyczący rozpatrywania skarg | Numer klauzuli odniesienia / artykuł dotyczący nadzoru / zgłaszania zdarzeń niepożądanych |
| Unia Europejska | EU MDR 2017/745 | art. 83 załącznik III (system PMS); art. 10 ust. 9 | Artykuły 87–92 (System nadzoru), załącznik III |
| Stany Zjednoczone (FDA) | 21 CFR część 820 (QSR) | §820.198 – Akta skarg | 21 CFR część 803 – System zgłaszania wyrobów medycznych (MDR) |
| Indie (CDSCO) | Przepisy dotyczące wyrobów medycznych z 2017 r. (MDR) | Rozdział VII, zasady 25 i 26 | Rozdział VII, zasada 27 |
| Globalne (norma ISO) | ISO 13485:2016 | Punkt 8.2.2 – Rozpatrywanie reklamacji | Punkt 8.2.3 – Sprawozdawczość wobec organów regulacyjnych |
Podsumowanie
W dzisiejszym konkurencyjnym środowisku regulacyjnym dotyczącym wyrobów medycznych skuteczne systemy zarządzania skargami i nadzoru rynkowego są absolutną koniecznością. Procesy te, zgodne z ISO 13485:2016, nie tylko chronią pacjentów, ale także przyczyniają się do budowania prężnej marki, której fundamentem jest reputacja. Inwestując w systematyczne procedury, ciągłe szkolenia oraz nadzór rynkowy po wprowadzeniu produktu do obrotu, producenci mogą przekształcić wymogi regulacyjne w szanse na rozwój i zdobycie pozycji lidera na rynku.









