
3 Min. Lesezeit
Ein Validierungsprotokoll ist ein dokumentierter Plan zur Prüfung eines Medizinprodukts, der bestätigt, dass der zur Herstellung des Produkts verwendete Produktionsprozess die spezifischen Anforderungen des Anwenders sowie technische und regulatorische Vorgaben erfüllt. Dies beinhaltet eine Überprüfung der Prozessvariablen und Betriebsgrenzen sowie die Analyse der Testergebnisse unter tatsächlichen Einsatzbedingungen.
Der Validierungsprozess umfasst mehrere konkrete Schritte. Diese werden wie folgt erläutert:
- Zuerst wird das Validierungsteam gebildet, und jedem Mitglied werden spezifische Rollen und Verantwortlichkeiten zugewiesen. Der Zweck der Prozessvalidierung ist es, die Validierungsziele klar zu formulieren und den Umfang der Validierungsaktivitäten festzulegen, indem die zu validierenden Aspekte des Medizinprodukts genau bestimmt werden. Anschließend erfasst das Team die grundlegenden Prinzipien des Prozesses, um spezifische Parameter und gewünschte Ergebnisse zu ermitteln.
- Zweitens werden die Bewertungs- und Akzeptanzkriterien festgelegt, zusammen mit der Auswahl geeigneter Testmethoden, Werkzeuge und statistischer Analysetechniken. Anschließend werden Prozessvalidierungsprotokolle entworfen und die Installationsqualifizierung (IQ), die Funktionsqualifizierung (OQ) sowie die Leistungsqualifizierung (PQ) durchgeführt.
- Zuletzt werden fortlaufende Prozesskontrollen und Überwachungsmaßnahmen festgelegt, um die kontinuierliche Validierung des Prozesses sicherzustellen. Bei Bedarf wird eine Revalidierung durchgeführt, um die Genauigkeit und Wirksamkeit des Validierungsprozesses aufrechtzuerhalten.
Abbildung 1 unten bietet eine Schritt-für-Schritt-Darstellung des Validierungsprozesses.
Abbildung 1: Die Phasen des Validierungsprozesses
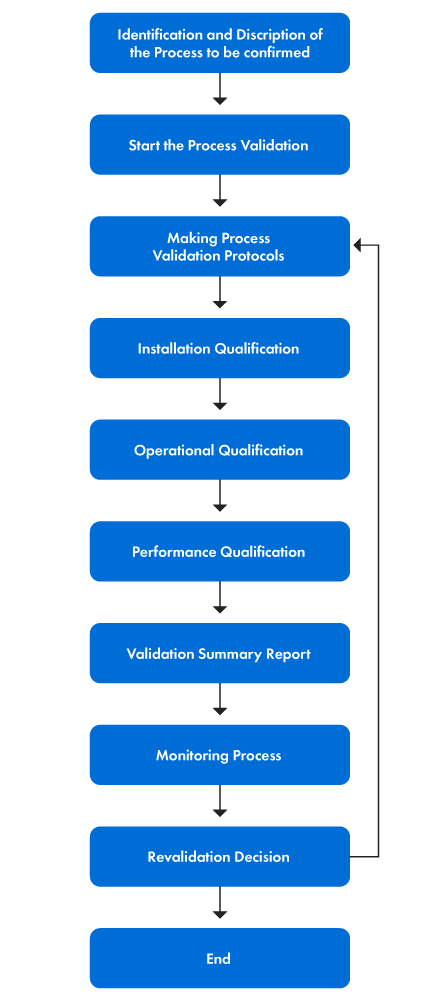
PVP
Aufgrund der großen Bandbreite an Produktionsvolumen und Fertigungskomplexitäten gibt es zahlreiche Ansätze zur Durchführung der Prozessvalidierung. Die Vorschriften der United States Food and Drug Administration (USFDA) und ISO 13485 bieten jedoch nur begrenzte Vorschläge zu spezifischen Methoden. Dennoch ist eine weit anerkannte und maßgebliche Quelle für die Prozessvalidierung von Medizinprodukten ein Leitfaden der Global Harmonization Task Force (GHTF), die heute als International Medical Device Regulators Forum (IMDRF) bekannt ist und 2004 veröffentlicht wurde. Er bleibt die primäre Referenz, selbst auf der offiziellen Website der USFDA.
Gemäß dem Leitfaden wird ein Validierungsteam gebildet, um einen detaillierten Prozessvalidierungsplan (PVP) zu erstellen. Prozessvalidierungsprotokolle enthalten ein detailliertes Schema zur Umsetzung von IQ, OQ, PQ und Revalidierung. Der PVP sollte die folgenden Elemente enthalten:
- Das Produkt definieren und den Validierungsansatz festlegen.
- Die Elemente identifizieren, die einer Validierung bedürfen.
- Aktivitäten am vorgesehenen Ort durchführen.
- Den Umfang der Dokumentation darlegen.
- Einen Zeitplan für die Validierungsaktivitäten erstellen.
- Erstellung eines Gesamtzeitplans.
- Pflege einer umfassenden Liste und Referenzen zu allen durchgeführten internen und externen Validierungen.
Das Validierungsprotokoll wird vor der Durchführung von Validierungsaktivitäten erstellt. Es sollte vom Validierungsteam vorbereitet und von der zuständigen Abteilung genehmigt werden. Der Zweck eines Validierungsprotokolls ist es, die Testskripte festzulegen, die befolgt werden müssen, um zu gewährleisten, dass die Prozesse und Geräte bereit sind, sichere und wirksame Medizinprodukte herzustellen.
Ein Analysebericht, der Informationen zusammen mit den notwendigen Analysen, Erläuterungen und Empfehlungen enthält, ist Teil des Validierungsprotokolls. Diese Aufzeichnungen werden zusätzlich überprüft, um sicherzustellen, dass die folgenden zwei (02) Kriterien erfüllt sind:
- Erfüllung regulatorischer Standards.
- Alle erzeugten Aufzeichnungen und Daten werden auf Ergebnisse, Angemessenheit und Vollständigkeit überprüft.
Abbildung 2 unten zeigt das PVP und die verschiedenen damit verbundenen Prozesse.
Abbildung 2: Das PVP und seine Anforderungen
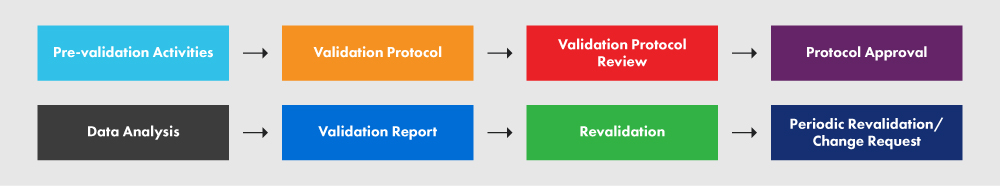
Ein entsprechend ausgearbeitetes Protokoll bietet klare Richtlinien, Vorgaben und Verfahren, die während der Prozessvalidierung einzuhalten sind. Es umfasst Aspekte wie Einrichtungen, Ausrüstung, Methoden und Schulungen. Das Protokoll legt die Prozesseingaben und -grenzen sowie die wesentlichen Schritte für die erfolgreiche Durchführung des Prozessvalidierungsprojekts fest. Obwohl die folgende Gliederung nicht jedes einzelne in Ihrem Protokoll erforderliche Element umfasst, gibt sie Ihnen einen Überblick über den erforderlichen Detaillierungsgrad. Wir empfehlen dringend, das Leitdokument zu befolgen, um ein besseres Verständnis des Prozesses zu erhalten.
- Titelseite
- Abzudeckende Produkte
- Zu validierende Ausrüstung/Prozess
- Allgemein
- Ziele
- Referenzdokumente
- Validierungsplan
- IQ
- OQ
- PQ
- Mess-/Prüfmittel und Kalibrierung
- Gerätewartung
- Revalidierung
- Genehmigungs-/Unterschriftsseite des Validierungsteams
Das Betriebsmanagement spielt eine entscheidende Rolle bei der Aufrechterhaltung optimaler Leistung, indem es Schlüsselkennzahlen überwacht, Arbeitsmethoden und -verfahren überprüft und bei auftretenden Problemen umgehend Maßnahmen ergreift. In Fällen, in denen Probleme auftreten, müssen Sie möglicherweise einen Prozess teilweise oder sogar vollständig neu validieren. Gemäß Abschnitt 820.75(c) der USFDA Quality System Regulation (QSR) sollte eine Prozessrevalidierung unter diesen Umständen in Betracht gezogen werden: „Wenn Änderungen oder Prozessabweichungen auftreten, muss der Hersteller diese überprüfen, bewerten und gegebenenfalls eine Revalidierung durchführen. Diese Aktivitäten müssen dokumentiert werden.“
Mögliche Auslöser für eine Prozessrevalidierung sind Änderungen an Spezifikationen, Methoden, Verfahren, Software, Designs, Schlüsselkomponenten, Chargengrößen, Standortwechseln, Geräteänderungen und Ähnlichem. Darüber hinaus kann die Implementierung von Korrektur- und Vorbeugemaßnahmen (CAPA) ebenfalls als Auslöser für eine Prozessrevalidierung dienen. Die Hauptgründe für eine Revalidierung sind folgende:
- Änderungen am Prozess.
- Negativer Qualitätstrend, plötzliche Qualitätsverschlechterung oder ein Anstieg von Kundenbeschwerden.
- Erhebliche Erweiterung der Linienkapazität.
- Designänderungen.
- Änderungen an der Produktverpackung.
- Verlagerung eines Prozesses an einen anderen Standort.
- Änderungen im Antragsverfahren.
Um mehr über Validierungsprotokolle und deren Bedeutung im Bereich der Medizinprodukteherstellung zu erfahren, kontaktieren Sie uns. Bleiben Sie informiert! Bleiben Sie konform!









