
Resumen de los servicios de cumplimiento del Reglamento sobre dispositivos médicos in vitro (IVDR) de la UE
El Reglamento europeo sobre productos sanitarios para diagnóstico in vitro (IVDR de la UE) constituye un nuevo marco normativo para la comercialización en el mercado europeo de los productos sanitarios para diagnóstico in vitro. El Reglamento IVDR de la UE está destinado a sustituir a la actual Directiva de la UE sobre productos sanitarios para diagnóstico in vitro (UE 98/79/CE). Al tratarse de un reglamento europeo, será aplicable en todos los Estados miembros de la UE y en los Estados miembros de la Asociación Europea de Libre Comercio (AELC), sin que sea necesario transponerlo al derecho de los Estados en cuestión. Como socio regulatorio de confianza, Freyr ofrece a las empresas servicios de cumplimiento normativo y asesoramiento en materia de IVDR dentro de la UE, con el fin de garantizar el cumplimiento de la normativa sobre diagnóstico in vitro y el marcado CE.
El Reglamento IVDR: calendario de transición
Prevista para entrar en vigor el 26 de mayo de 2022, la nueva normativa europea IVDR se considera un cambio significativo en la supervisión reglamentaria de los productos sanitarios para diagnóstico in vitro. El resumen de la transición al IVDR es el siguiente:
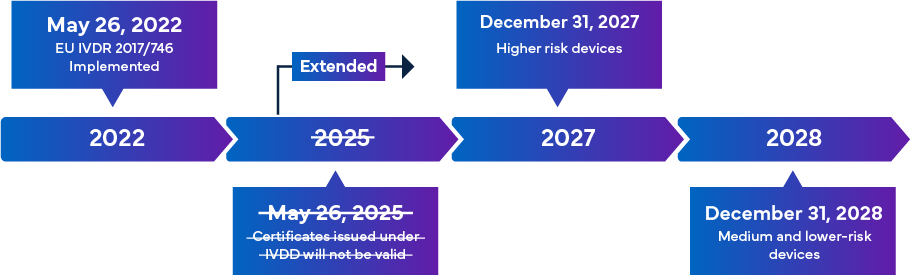
Una vez que se hayan aplicado plenamente, las normas de cumplimiento del IVDR garantizan que casi todos los productos para diagnóstico in vitro (IVD) que entren en el mercado europeo se sometan al examen del organismo notificado (ON) del IVDR y a la certificación del marcado CE en el marco del procedimiento de autorización de los IVD. En este escenario, además de la adaptación a la clasificación actualizada del IVDR, los fabricantes deben revisar inmediatamente la documentación técnica clave para completar con éxito el registro del IVD y el marcado CE. Los requisitos del IVDR varían en función de la clase de riesgo del IVD. En general, para la certificación de los IVD, los fabricantes deben realizar las siguientes operaciones:
- Revisión de los expedientes técnicos de conformidad con el Reglamento IVDR (Reglamento (UE) 2017/746)
- Elaboración del informe de evaluación del rendimiento (PER) para todos los productos de diagnóstico in vitro
- Seguimiento del rendimiento tras la comercialización (PMPF) de conformidad con el anexo XIII, parte B, del IVDR.
- Informe de vigilancia de conformidad con el artículo 82 del IVDR
Freyr ayuda a sus clientes a llevar a cabo una revisión sistemática de la literatura científica y les asiste en la planificación y preparación de un PER para cumplir con la normativa sobre productos de diagnóstico in vitro. Freyr cuenta con expertos especializados que prestan servicios de asesoramiento en materia de IVDR y apoyo a la vigilancia poscomercialización (SPM), que forma parte integrante del sistema de gestión de la calidad del fabricante, así como al seguimiento del rendimiento poscomercialización (SPPM).
Obtenga asesoramiento de expertos sobre el cumplimiento del Reglamento IVDR de la UE
Servicios de cumplimiento del Reglamento IVDR de la UE: LA EXPERIENCIA Y LAS VENTAJAS DE
- Plan de transición para el cumplimiento del Reglamento sobre dispositivos médicos in vitro (IVDR)
- Análisis técnico y evaluación de las deficiencias de los requisitos del IVDR en relación con los GSPR (requisitos generales de seguridad y rendimiento)
- Apoyar la elaboración del expediente técnico de conformidad con los requisitos del IVDR
- Informes de validez científica basados en la bibliografía y/o en datos internos
- Informes sobre el rendimiento clínico basados en la bibliografía y/o en datos internos
- Datos clínicos o informes de evaluación del rendimiento
- Protocolos e informes de seguimiento del rendimiento poscomercialización (SPPM)
- Protocolos e informes de vigilancia poscomercialización (SPM)
- Redactar o revisar otros documentos, como prospectos o instrucciones de uso (IFU), guías de referencia rápida (QRI), manuales de usuario o de funcionamiento, etc.

- Garantizar el cumplimiento del Reglamento sobre productos sanitarios in vitro (IVDR), el registro de productos sanitarios in vitro (IVD) y el marcado CE
- Profundo conocimiento de la normativa y experiencia en los ámbitos clave de aplicación del Reglamento sobre dispositivos médicos de la UE (IVDR)
- Modelo de entrega centrado en la gestión de proyectos para garantizar el cumplimiento del calendario.
- Expertos internos de la ON (revisión del informe por parte de los evaluadores interactivos de la ON)
- Equipos especializados con conocimientos interdisciplinarios en ámbitos de impacto y categorías de dispositivos específicos
- Aportaciones interdisciplinarias de expertos en productos sanitarios para cumplir con los requisitos
- Conjunto de servicios relacionados con el cumplimiento normativo, la revisión y la planificación
- Gran experiencia en garantizar la coherencia de los resultados (plazos y calidad)
