
Cos'è il QMSR?
FDA rappresenta un approccio semplificato volto ad allinearsi ai requisiti ISO 13485:2016, che costituisce un aggiornamento della precedente struttura QSR. Questo allineamento è fondamentale perché semplifica la conformità normativa per i produttori quelli che operano a livello globale. Tale armonizzazione consentirà alle aziende di soddisfare i requisiti normativi sia negli US in altri mercati in modo molto più uniforme.
Il QMSR impone miglioramenti nella gestione dei rischi, nella progettazione dei dispositivi e nella sorveglianza post-commercializzazione. Questo aggiornamento potrebbe comportare una maggiore complessità, ma offre anche ai produttori standardizzare le proprie procedure di qualità, il che a sua volta aumenta la sicurezza dei dispositivi e garantisce una documentazione più accurata, che può rivelarsi fondamentale durante USFDA .
Perché il passaggio da QSR a QMSR è fondamentale?
![]()
![]()
Conformità agli standard internazionali
Ciò aiuterà i produttori allinearsi al sistema di gestione della qualità (SGQ) globale per i requisiti relativi ai dispositivi medici, passando al QMSR, il che riduce le sovrapposizioni normative per i produttori di dispositivi medici produttori a livello globale.
![]()
Maggiore qualità, sicurezza ed efficacia
Questa integrazione garantisce che i dispositivi siano prodotti in conformità con i requisiti globali del sistema di gestione della qualità. Ciò aiuta il produttore a migliorare la qualità e la sicurezza dei dispositivi.
![]()
Conformità Regolatoria
Ciò aiuta i produttori adeguarsi ai nuovi requisiti e riduce il rischio di non conformità quando il regolamento entrerà in vigore il 2 febbraio 2026.



Principali modifiche relative al passaggio da QSR a QMSR:
![]()
Allineamento alla terminologia della ISO 13485:2016
Ciò garantisce che produttori standard riconosciuti a livello mondiale.
![]()
Gestione dei rischi:
Pone l'accento sulla gestione dei rischi durante l'intero ciclo di vita del dispositivo medico.
![]()
Progettazione e comandi del dispositivo
Ai sensi del FDA , i controlli di progettazione sono stati ampliati per garantire che produttori tengano produttori conto delle esigenze degli utenti, della sicurezza del dispositivo e dei criteri di prestazione, aspetti che costituiscono un’area di particolare attenzione durante USFDA
![]()
Sorveglianza post-commercializzazione.
Le aziende devono potenziare il sistema di monitoraggio post-commercializzazione. Ciò richiederà produttori raccogliere informazioni sulla sicurezza e sull'efficacia dei dispositivi, al fine di individuare e risolvere rapidamente eventuali problemi.
![]()
Documentazione e tenuta dei registri
Il regolamento definitivo sottolinea l'importanza di una documentazione completa, poiché la tenuta accurata dei registri è fondamentale durante l'ispezione.
Date importanti
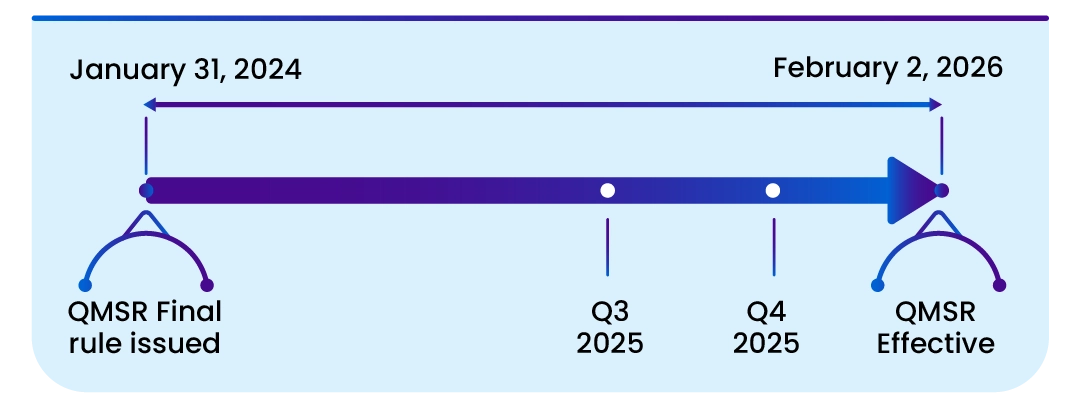
La presente norma entra in vigore il 2 febbraio 2026. L'inserimento per riferimento di alcuni documenti elencati nella presente norma è stato approvato dal Direttore del Registro Federale il 2 febbraio 2024.
Ottieni una consulenza specialistica sulla conformità al QMSR
Perché è importante attenersi al quadro normativo FDA ?
![Intelligence Normativa (RI)]() Il QMSR entrerà in vigore il 2 febbraio 2026; produttori saranno tenuti produttori adeguare il proprio sistema di gestione della qualità.
Il QMSR entrerà in vigore il 2 febbraio 2026; produttori saranno tenuti produttori adeguare il proprio sistema di gestione della qualità.![Un unico partner per tutto.]() Un'implementazione puntuale e una manutenzione efficace possono ridurre il rischio di richiami dei prodotti e di reclami.
Un'implementazione puntuale e una manutenzione efficace possono ridurre il rischio di richiami dei prodotti e di reclami.![garantire l'accuratezza della sottomissione]() Il rispetto delle norme QMSR potrebbe ridurre il numero di osservazioni da parteFDA US FDA senzaFDA incida sullo status del prodotto nel mercato regolamentato.
Il rispetto delle norme QMSR potrebbe ridurre il numero di osservazioni da parteFDA US FDA senzaFDA incida sullo status del prodotto nel mercato regolamentato.
 Il QMSR entrerà in vigore il 2 febbraio 2026; produttori saranno tenuti produttori adeguare il proprio sistema di gestione della qualità.
Il QMSR entrerà in vigore il 2 febbraio 2026; produttori saranno tenuti produttori adeguare il proprio sistema di gestione della qualità. Un'implementazione puntuale e una manutenzione efficace possono ridurre il rischio di richiami dei prodotti e di reclami.
Un'implementazione puntuale e una manutenzione efficace possono ridurre il rischio di richiami dei prodotti e di reclami. Il rispetto delle norme QMSR potrebbe ridurre il numero di osservazioni da parteFDA US FDA senzaFDA incida sullo status del prodotto nel mercato regolamentato.
Il rispetto delle norme QMSR potrebbe ridurre il numero di osservazioni da parteFDA US FDA senzaFDA incida sullo status del prodotto nel mercato regolamentato.Scegliete i servizi di consulenza FDA offerti da Freyr: i nostri consulenti, tra i migliori del settore, vi accompagneranno con la massima cura in ogni fase del ciclo di vita del vostro dispositivo, per garantire un'attuazione senza intoppi dei requisiti QMSR!
Ottieni una consulenza specialistica sulla conformità al QMSR
QMSR
- Classificazione dei dispositivi medici secondo laFDA.
- Creazione di documenti in conformità al 21 CFR 820.
- Analisi delle discrepanze dei documenti esistenti del Sistema di gestione della qualità (SGQ) ai sensi del 21 CFR 820.
- Piano di bonifica per la conformità a 21 CFR 820.
- Simulazioni di audit.
- Un team dedicato di esperti di Garanzia della Qualità (QA) sui dispositivi medici.
- Comprovata esperienza nella gestione della conformità a 21 CFR 820.
- Modelli flessibili di erogazione dei progetti.