
5 min czytania
Oprogramowanie wyrobów medycznych w Korei Południowej jest wykorzystywane do diagnozowania, leczenia i monitorowania pacjentów w nowoczesnym systemie opieki zdrowotnej. Obejmuje ono zarówno oprogramowanie wbudowane, zintegrowane z wyrobami medycznymi, jak i oprogramowanie samodzielne, które może być używane na komputerach PC, urządzeniach mobilnych i w usługach internetowych. Ministerstwo Bezpieczeństwa Żywności i Leków (MFDS) Korei Południowej odpowiada za regulację oprogramowania wyrobów medycznych oraz zapewnienie jego bezpieczeństwa i skuteczności. 5 lipca 2023 r. MFDS określiło kryteria zatwierdzania i kontroli oprogramowania wyrobów medycznych; przepisy te stanowią strukturę, którą wnioskodawcy mogą stosować przy składaniu oprogramowania do zatwierdzenia lub przeglądu.
Przepisy dotyczą różnorodnych tematów, w tym zakresu zastosowania, wymagań dotyczących dokumentacji technicznej oraz raportów weryfikacji zgodności. Istnieją międzynarodowe normy i wytyczne, które mają zastosowanie do oprogramowania wyrobów medycznych, oprócz wytycznych MFDS, takie jak norma Międzynarodowej Komisji Elektrotechnicznej (IEC) 62304 dotycząca procesów cyklu życia oprogramowania oraz wytyczne Amerykańskiej Agencji ds. Żywności i Leków (US FDA) dotyczące mobilnych aplikacji medycznych.
Plan rozwoju oprogramowania i analiza wymagań
- Plan rozwoju oprogramowania przedstawia ogólne podejście do rozwoju oprogramowania, w tym specyfikacje, metody i narzędzia rozwojowe. Obejmuje również weryfikację, zarządzanie ryzykiem wyrobów medycznych, zarządzanie konfiguracją i dokumentację.
- Analiza wymagań określa wymagania dotyczące oprogramowania wyrobów medycznych, w tym środki kontroli ryzyka i metody weryfikacji. Poprzez staranne planowanie i analizowanie procesu tworzenia oprogramowania, deweloperzy mogą zapewnić, że powstałe oprogramowanie spełnia niezbędne standardy bezpieczeństwa i skuteczności.
- Raport weryfikacji zgodności oprogramowania zawiera zarys planu rozwoju oprogramowania, numer kontrolny dokumentu producenta oraz przegląd analizy wymagań. Przestrzeganie tych wytycznych pozwala na pewne tworzenie oprogramowania dla wyrobów medycznych, z gwarancją, że przeszło ono rygorystyczne testy i spełnia niezbędne normy bezpieczeństwa i skuteczności.
Weryfikacja i walidacja oprogramowania wyrobów medycznych
- Weryfikacja oprogramowania wyrobów medycznych zapewnia, że oprogramowanie spełnia określone wymagania.
- Walidacja oprogramowania wyrobów medycznych zapewnia, że oprogramowanie spełnia potrzeby użytkownika i zamierzone zastosowania.
- Raport z weryfikacji i walidacji oprogramowania wyrobu medycznego przedstawia proces weryfikacji i walidacji, w tym nazwę produktu, wersję oraz nazwiska osób, które zbadały i zatwierdziły raport. Raport może się różnić w zależności od charakterystyki oprogramowania, ale powinien zawierać opis oprogramowania, zastosowane metody weryfikacji i walidacji oraz wyniki testów.
Środowisko operacyjne i oprogramowanie nieznanego pochodzenia (SOUP)
- Jeśli oprogramowanie jest zależne od określonego sprzętu, np. oprogramowania wbudowanego, dokumentacja techniczna powinna opisywać specyfikacje sprzętowe.
- Jednakże, jeśli oprogramowanie jest samodzielne i opracowane do działania na sprzęcie ogólnego przeznaczenia, środowisko operacyjne musi być opisane w dokumentacji technicznej. Obejmuje to minimalne zalecane specyfikacje, takie jak Microsoft Windows 10 lub nowszy.
- Ponadto, jeśli oprogramowanie wyrobu medycznego zawiera komercyjne oprogramowanie nieznanego pochodzenia (SOUP), należy stworzyć środowisko operacyjne, aby zapewnić jego prawidłowe działanie. Poprzez dokładne opisanie środowiska operacyjnego i uwzględnienie wszelkich SOUP, programiści mogą zapewnić, że ich oprogramowanie wyrobu medycznego jest bezpieczne i skuteczne do zamierzonego zastosowania.
Zarządzanie ryzykiem wyrobów medycznych i wymagania dotyczące dokumentacji
- Proces zarządzania ryzykiem oprogramowania jako wyrobu medycznego obejmuje identyfikację sytuacji niebezpiecznych, ustanawianie środków kontroli ryzyka, weryfikację tych środków oraz zarządzanie zmianami w oprogramowaniu.
- Dokument MFDS-RM dotyczący zarządzania ryzykiem oprogramowania zawiera informacje na temat zarządzania ryzykiem związanym z oprogramowaniem.
- Ponadto wymogi dotyczące dokumentacji są kluczowe, aby zapewnić, że oprogramowanie spełnia niezbędne standardy bezpieczeństwa i skuteczności.
- Plan rozwoju oprogramowania, analiza wymagań oprogramowania wyrobu medycznego oraz raporty z weryfikacji i walidacji oprogramowania muszą być zawarte w dokumentacji.
- Raport z weryfikacji zgodności oprogramowania przedstawia wymagania dotyczące dokumentacji; zawiera również zarys obowiązujących dokumentów oraz numer kontrolny dokumentu producenta.
Rysunek 1: Proces zarządzania ryzykiem wyrobów medycznych
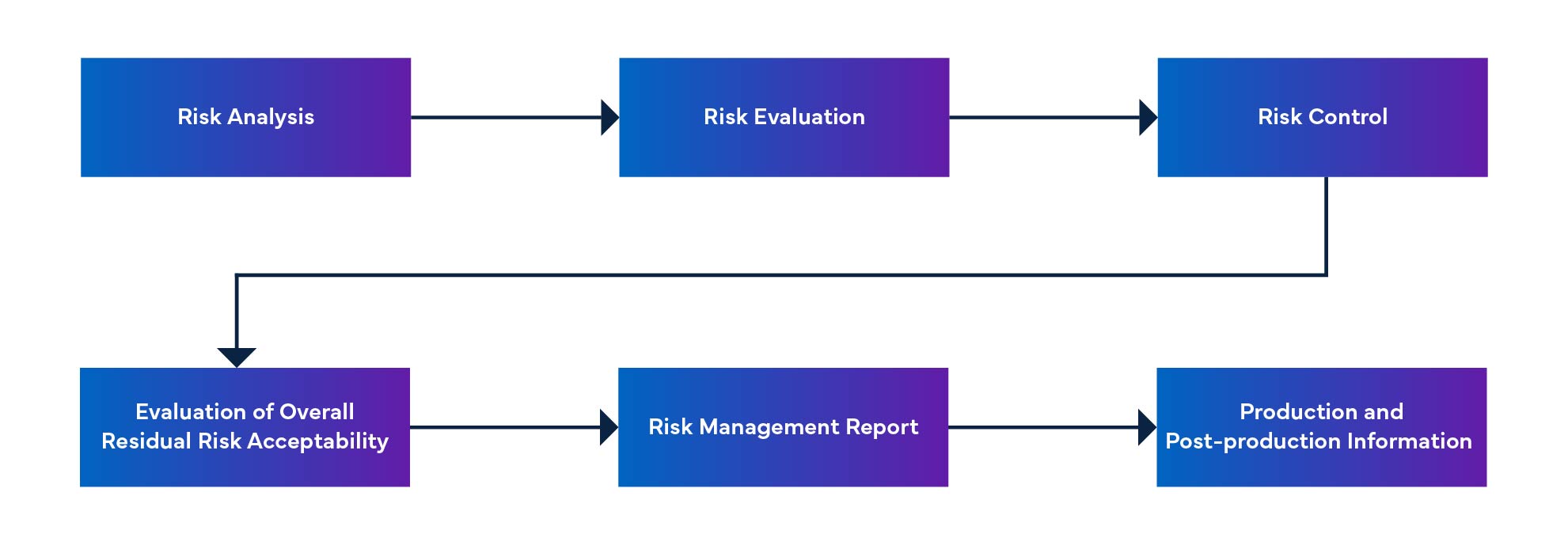
Nierozwiązane anomalie i działania korygujące dla oprogramowania SaMD
- Dokument MFDS-PR (Rozwiązywanie Problemów Oprogramowania) przedstawia proces rozwiązywania problemów z oprogramowaniem, który obejmuje zgłaszanie problemów, analizę, wdrożenie i weryfikację.
- Dokument zawiera również listę nierozwiązanych problemów, błędów, usterek i anomalii, a także ocenę ryzyka resztkowego dla systemu oprogramowania.
- Działania naprawcze podjęte w celu rozwiązania tych problemów muszą być udokumentowane w planie konserwacji oprogramowania, który jest ustalany zgodnie z procesem konserwacji oprogramowania.
- Dokument MFDS dotyczący utrzymania zawiera informacje na temat konserwacji i rozwiązywania problemów z oprogramowaniem SaMD.
Przegląd dokumentacji technicznej i wymagania dotyczące składania dokumentów dla oprogramowania SaMD.
Głównymi dokumentami przeglądowymi podczas procesu oceny są dane dotyczące wydajności, raport potwierdzenia zgodności oraz dane z weryfikacji i walidacji oprogramowania wyrobu medycznego, specyfikacja projektu oprogramowania (SDS), oświadczenie o wymaganiach oprogramowania wyrobu medycznego (SRS) oraz raporty z weryfikacji i walidacji. Raport potwierdzenia zgodności oraz Raport z weryfikacji i walidacji oprogramowania wyrobu medycznego muszą zostać złożone.
Zarządzanie ryzykiem w oprogramowaniu wyrobów medycznych
- Identyfikacja potencjalnych zagrożeń związanych z oprogramowaniem i jego użytkowaniem.
- Ocena stopnia ryzyka związanego z tymi zagrożeniami.
- Wdrażanie środków kontroli ryzyka w celu zminimalizowania prawdopodobieństwa szkody.
- Monitorowanie i przegląd skuteczności tych środków kontroli ryzyka.
- Dokumentowanie wszystkich działań i decyzji związanych z zarządzaniem ryzykiem wyrobów medycznych.
W systemie oprogramowania, elementy oprogramowania są dzielone na mniejsze części, w tym szczegółowe elementy oprogramowania. Gdy elementu nie można już dalej dzielić, nazywany jest on jednostką. System umożliwia podział do poziomu jednostki, pomagając określić poziom bezpieczeństwa dla każdego elementu oprogramowania. Łącząc te elementy oprogramowania, jesteśmy w stanie określić poziom bezpieczeństwa dla całego systemu oprogramowania.
Rysunek 2: Demontaż i integracja oprogramowania wyrobów medycznych
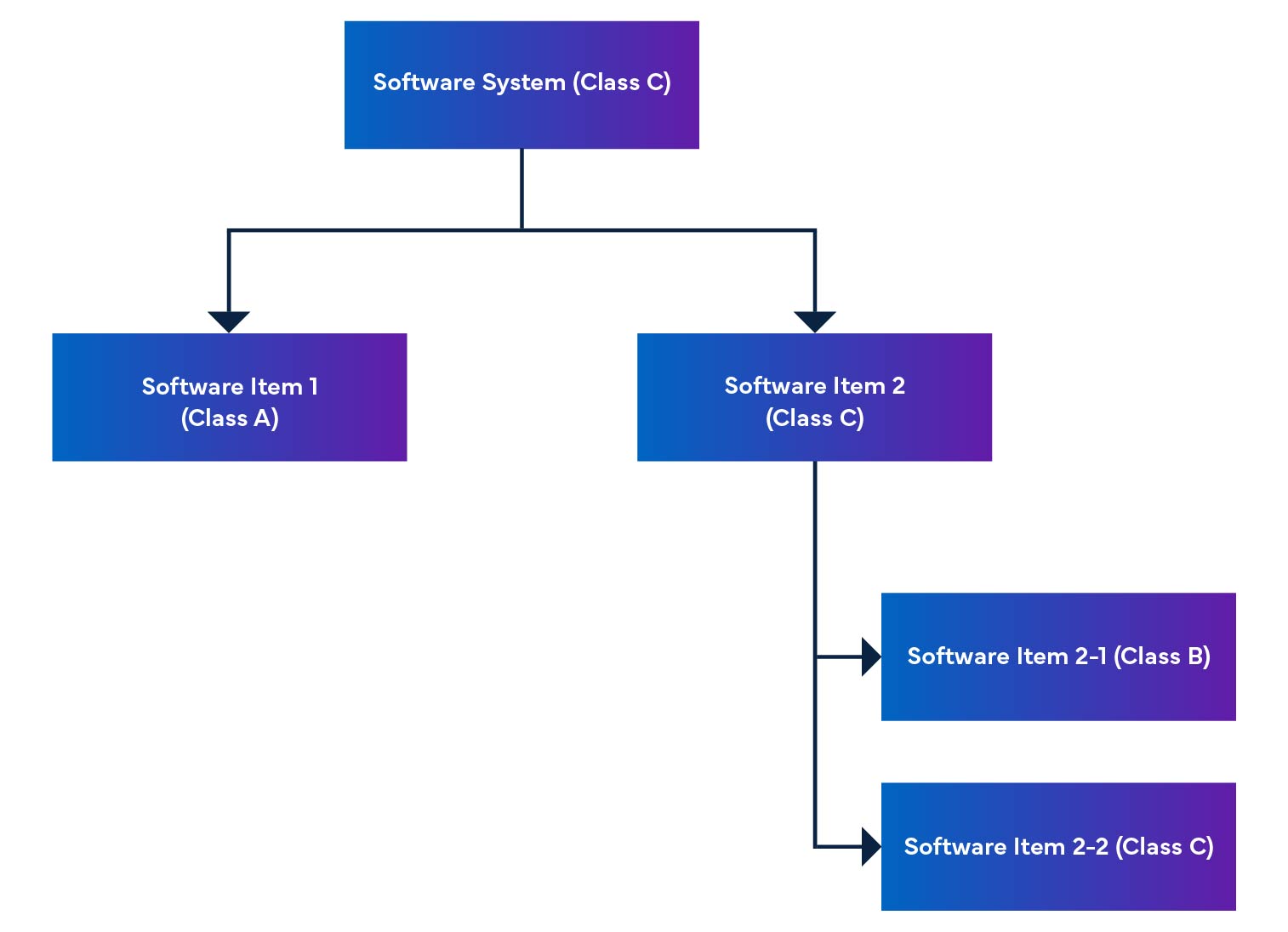
Rozporządzenie wspomina również o ocenie bezpieczeństwa oprogramowania, która służy do identyfikacji ryzyka związanego z oprogramowaniem SaMD (patrz Tabela 1).
Tabela 1: Definicja oceny bezpieczeństwa
| Ocena | Definicja klasy bezpieczeństwa oprogramowania wyrobów medycznych |
| Klasa A | Brak możliwości urazu lub uszkodzenia ciała. |
| Klasa B | Prawdopodobne są mniej poważne urazy (drobne urazy). |
| Klasa C | Możliwość poważnych obrażeń lub śmierci. |
Zarządzanie konfiguracją oprogramowania
- Utrzymywanie dokładnej i aktualnej dokumentacji dla wszystkich wersji oprogramowania, zmian i aktualizacji.
- Zapewnienie, że cała dokumentacja jest odpowiednio przeglądana i zatwierdzana.
- Wdrażanie procedur zarządzania zmianami konfiguracji oprogramowania.
- Dokumentowanie wszystkich działań i decyzji związanych z zarządzaniem konfiguracją oprogramowania.
Utrzymanie oprogramowania
- Regularne testowanie i monitorowanie oprogramowania w celu zapewnienia, że pozostaje ono bezpieczne i skuteczne zgodnie z przeznaczeniem.
- Wdrażanie procedur w celu rozwiązywania wszelkich problemów, które mogą się pojawić, w tym poprawek błędów i aktualizacji oprogramowania.
- Dokumentowanie wszystkich działań i decyzji związanych z utrzymaniem oprogramowania.
Rozwiązywanie problemów
- Identyfikacja pierwotnej przyczyny problemu.
- Wdrażanie działań naprawczych w celu rozwiązania problemu.
- Dokumentowanie całego procesu rozwiązywania problemów do wykorzystania w przyszłości.
Postępując zgodnie z powyższymi wytycznymi, deweloperzy mogą zapewnić, że wszelkie problemy z oprogramowaniem ich wyrobów medycznych zostaną odpowiednio rozwiązane i udokumentowane, a oprogramowanie spełnia niezbędne wymagania do zatwierdzenia lub badania.
Jeśli jesteś producentem wyrobów medycznych dążącym do zgodności z południowokoreańskimi normami oprogramowania wyrobów medycznych, eksperci Freyr ds. regulacji mogą Cię poprowadzić przez złożone środowisko regulacyjne tego kraju. Zapewnimy, że Twoje urządzenia są zgodne z najnowszymi przepisami dotyczącymi wyrobów medycznych w Korei Południowej, co zapewni bezproblemową zgodność. Skontaktuj się z nami, aby dowiedzieć się więcej!









