
2 minuty czytania
Wyroby medyczne przechodzą ocenę zgodności, zanim zostaną włączone do listy ARTG, aby upewnić się, że wyroby są zgodne z podstawowymi zasadami wymaganymi przez Therapeutic Goods Administration (TGA) w Australii. Podstawowe zasady zasadniczo określają charakterystykę bezpieczeństwa i działania, którą każdy wyrób powinien spełniać, aby mógł być sprzedawany w Australii. Podobnie jak w UE, ocena zgodności w Australii opiera się na klasie ryzyka wyrobu. Dlatego ścieżka oceny zgodności, którą należy zastosować dla danego wyrobu, zależy od jego klasyfikacji.
Istnieją różne standardy regulacyjne, które mają zastosowanie do różnych rodzajów wyrobów. Zgodność z jednym lub kilkoma z tych standardów jest wymagana jako zasadniczy wymóg. Identyfikacja odpowiednich standardów mających zastosowanie do wyrobu oraz późniejsze testowanie wyrobu w celu wykazania jego zgodności ze standardem staje się warunkiem wstępnym.
Ocena zgodności obejmuje systematyczne badanie dokumentów technicznych dotyczących wyrobu. Zarządzanie ryzykiem, ocena kliniczna, zaangażowany proces produkcyjny oraz działania w zakresie nadzoru prowadzone przez producenta to kluczowe obszary oceny. Wyroby, które muszą posiadać certyfikat oceny zgodności TGA, są wymienione w Rozporządzeniu 4.1 Therapeutic Goods (Medical Devices) Regulations 2002. Chociaż certyfikat oceny zgodności wydany przez TGA jest warunkiem wstępnym do wprowadzenia większości wyrobów medycznych w Australii, TGA akceptuje również certyfikaty oceny zgodności wydane przez Jednostki Notyfikowane w Europie. Ponadto TGA akceptuje również oceny zgodności z krajów będących częścią MDSAP (Medical Device Single Audit Program).
Wymogi oceny zgodności różnią się w zależności od kategorii ryzyka wyrobu. Tabela nr 1 szczegółowo przedstawia proces oceny zgodności w Australii, oparty na klasyfikacji.
Proces oceny zgodności - Australia
Klasa | Procedura oceny zgodności | Obowiązki producenta |
| I | Część 6 (Deklaracja zgodności, niewymagająca oceny przez Sekretarza) | Dokumentacja potwierdzająca zgodność z Zasadniczymi Wymaganiami |
| I (pomiarowe) i IIa (niesterylne) | Część 6 (Deklaracja zgodności, niewymagająca oceny przez Sekretarza) Część 5 (System zarządzania jakością produktu) | Dokumentacja potwierdzająca zgodność z Zasadniczymi Wymaganiami. Wdrożyć system zarządzania jakością produktu do końcowej kontroli i testowania na potrzeby audytów. Uwaga: Złożenie Deklaracji Zgodności nie jest wymagane dla Klasy I (niepomiarowe i niesterylne), ale powinno być dostępne na żądanie TGA. |
| I (sterylne) i IIa (sterylne) | Część 6 (Deklaracja zgodności, niewymagająca oceny przez Sekretarza) Część 4 (Zapewnienie jakości produkcji) | Dokumentacja potwierdzająca zgodność z Zasadniczymi Wymaganiami. Wdrażanie systemu zarządzania jakością z wyłączeniem elementu projektowego, zgodnie z ISO 13485. |
| IIb | Część 1 (Pełne zapewnienie jakości) z wyłączeniem klauzuli 1.6 (Badanie projektu) | Wdrażanie kompletnego systemu zarządzania jakością, obejmującego projektowanie, produkcję, etykietowanie, pakowanie i kontrolę końcową, zgodnie z ISO 13485. Z wyłączeniem dokumentacji projektowej. |
| III & AIMD | Część 1 (Pełne zapewnienie jakości) z uwzględnieniem klauzuli 1.6 (Badanie projektu) | Wdrożyć pełny system zarządzania jakością obejmujący projektowanie, produkcję, etykietowanie, pakowanie i kontrolę końcową, zgodnie z ISO 13485. Dokumentacja projektowa zgodnie z Zasadami Zasadniczymi. |
| Zestawy lub pakiety zabiegowe, Część 7 | (Procedury dotyczące wyrobów medycznych używanych do celów specjalnych) | Zasadnicze Wymagania. Procedura oceny zgodności. Dowody kliniczne dla poszczególnych komponentów w systemie lub opakowaniu. |
Tabela nr 1. Wymagania dotyczące oceny zgodności wyrobów
Zwykły proces zatwierdzania wyrobów medycznych przez TGA wygląda następująco:
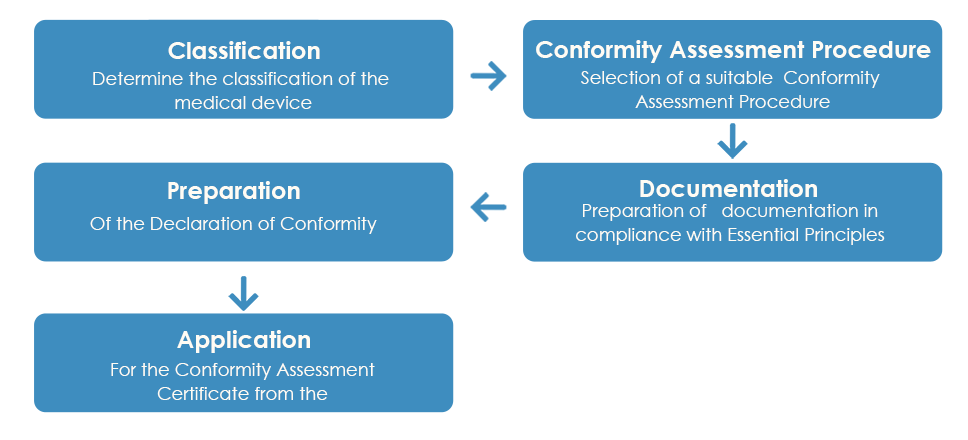
Jak szczegółowo opisano powyżej, po otrzymaniu Certyfikatu Oceny Zgodności od TGA, producent musi przygotować Deklarację Zgodności (DoC), oświadczającą, że wyrób medyczny jest zgodny z obowiązującymi zasadami zasadniczymi, zasadami klasyfikacji i drogą oceny zgodności. Jednakże, w przeciwieństwie do Certyfikatu Oceny Zgodności, europejska DoC nie jest akceptowana przez TGA.
Rynek australijski oferuje obiecujące perspektywy dla producentów wyrobów medycznych, jeśli spełnione zostaną wymagania regulacyjne Therapeutic Goods Administration (TGA). Wymagania dotyczące zgodności i proces oceny różnią się w zależności od klasy ryzyka wyrobu i IVD. Chociaż przepisy dotyczące wyrobów medycznych są dobrze zdefiniowane i przejrzyste, poruszanie się po nich jest złożone, a producenci mogą szukać wsparcia u partnerów regulacyjnych w celu pomyślnego wprowadzenia wyrobu na listę.
Aby uzyskać kompleksowe informacje na temat wymagań oceny zgodności, Australijskiego Sponsora i wpisu do ARTG w Australii, więcej informacji uzyskasz, kontaktując się ze sprawdzonym ekspertem ds. Spraw regulacyjnych. Bądź na bieżąco. Zachowaj zgodność.









