

4 min de lectura
Con la Administración de Alimentos y Medicamentos de los US (USFDA) implementando la regla final para las regulaciones del sistema de gestión de calidad (QMSR) en 2024, los fabricantes de Dispositivos Médicos deben adoptar las enmiendas para comercializar y distribuir sus dispositivos en el mercado de los US.
Esta norma actualiza los Reglamentos del Sistema de Calidad (QSR) de la USFDA al alinearse con la ISO 13485:2016, que es el estándar internacional para el QMS de Dispositivos Médicos. Los fabricantes de Dispositivos Médicos tienen un período de transición de dos años para adaptarse, lo que hace necesario que las organizaciones adapten los nuevos requisitos para evitar el incumplimiento en el momento de la inspección.
¿Qué es la QMSR?
La FDA QMSR es un enfoque simplificado para los requisitos de QMS, que es una actualización de la antigua estructura QSR. Esta alineación es crucial porque simplifica el cumplimiento global para los fabricantes, especialmente aquellos que operan a nivel global. Esta armonización permitirá a las empresas cumplir con los requisitos reglamentarios tanto en los US como en otros mercados de una manera mucho más unificada.
El Reglamento de Sistemas de Gestión de Calidad (QMSR) exige mejoras en la gestión de riesgos, el diseño de los dispositivos y la vigilancia post-comercialización. Este rol puede añadir más complejidad, pero también permite a los fabricantes estandarizar sus procedimientos de calidad, lo que a su vez aumenta la seguridad del dispositivo y mejora la documentación, lo cual puede ser vital durante las inspecciones de la USFDA.
Cambios clave en el QMSR
- Armonización con ISO 13485: Este es el paso más crucial que permite a los fabricantes de Dispositivos Médicos aceptar estándares reconocidos internacionalmente. La USFDA reconoció que muchas empresas de Dispositivos Médicos ya cumplen con la ISO 13485, lo que reduce los esfuerzos duplicados.
- La gestión de riesgos enfatiza la gestión de riesgos a lo largo de todo el ciclo de vida del Dispositivo Médico. Los fabricantes de Dispositivos Médicos deben demostrar una gestión de riesgos, un control de riesgos y una gestión de riesgos eficaces.
- Diseño y controles de dispositivos: Bajo el QMSR, los controles de diseño se ampliaron para asegurar que los fabricantes de Dispositivos Médicos tengan plenamente en cuenta las necesidades del usuario, la seguridad del dispositivo y los criterios de rendimiento, lo cual es un área de enfoque durante la inspección de la USFDA.
- Post Market Surveillance: Las empresas necesitan mejorar el sistema de monitoreo post-comercialización. Esto implicará que los fabricantes recopilen información sobre la seguridad y eficacia del dispositivo, lo que ayuda a encontrar e identificar los problemas rápidamente.
- Documentación y mantenimiento de registros: Esta es la norma final que hace hincapié en la documentación. Un mantenimiento de registros exhaustivo y adecuado es fundamental durante la inspección.
Pasos para prepararse para una inspección de la Administración de Alimentos y Medicamentos de US:
Dado que la inspección dura hasta 2026, las empresas tienen dos años para que sus sistemas de calidad se alineen con el QMSR. Sin embargo, esperar hasta el último minuto puede ser arriesgado.
Pasos para la inspección de la USFDA bajo el marco QMSR para industrias donde el SGC actual se basa en QSR:
- Realizar un análisis de brechas: Este es el primer paso en el que el sistema de calidad actual difiere de los nuevos requisitos del QMSR. Un análisis de brechas exhaustivo ayudará a identificar las áreas que necesitan actualizaciones, como la gestión de riesgos, la vigilancia post-comercialización y los controles de diseño.
- Actualizar el procedimiento de gestión de riesgos: Asegúrese de que las actividades de gestión de riesgos se combinen con todo el ciclo de vida de su producto, desde el diseño hasta la vigilancia post-comercialización.
- Revisar el control del diseño: Los fabricantes deben asegurarse de que el proceso de diseño sea sólido y esté bien documentado. Valide que el proceso de diseño sea sólido, esté bien documentado y esté completamente integrado en su sistema de gestión de calidad.
- Mejorar la vigilancia poscomercialización: Implementar sistemas para supervisar el rendimiento del dispositivo una vez que se comercializa. Esto podría implicar establecer mecanismos de retroalimentación de los clientes, recopilar evidencia de datos clínicos y realizar un seguimiento exhaustivo.
- Formación y documentación: Capacitación del personal sobre los nuevos requisitos, especialmente aquellos involucrados en la gestión de calidad y el cumplimiento reglamentario. Esto asegura que todo el proceso de documentación esté alineado con las expectativas de QMSR.
- Certificación de terceros: Si su empresa no cuenta con la certificación ISO 13485, ahora podría ser el momento de considerarla. Obtener la certificación ISO 13485 puede darle una ventaja para cumplir los requisitos de la USFDA y aumentar la credibilidad en los mercados globales.
Gestionando el período de transición de dos años
Para utilizar eficazmente el período de transición y garantizar el cumplimiento de la Regulación del Sistema de Gestión de Calidad (QMSR) de la USFDA, los fabricantes deben adoptar un enfoque proactivo. Aquí hay una hoja de ruta sugerida:
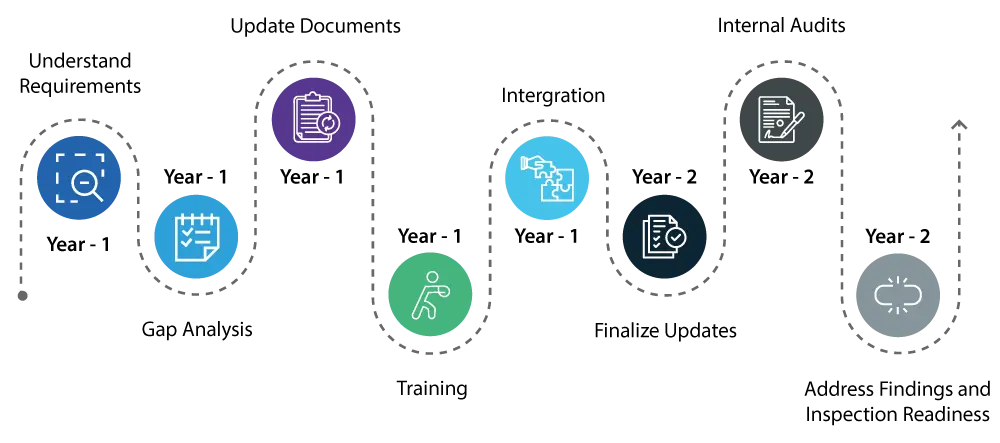
Año 1:
- Comprender el requisito: Empiece por comprender a fondo los cambios introducidos por el QMSR. Esto implica una revisión detallada de los nuevos requisitos y cómo difieren del Reglamento del Sistema de Calidad (QS) existente.
- Conozca las deficiencias: Realice un análisis exhaustivo de las deficiencias para identificar las áreas dentro de su sistema de calidad actual que requieren actualizaciones para cumplir con los nuevos estándares del QMSR.
- Actualización / subsanación de documentos: Inicie las actualizaciones necesarias en su sistema de calidad, centrándose en áreas como la gestión de riesgos y los controles de diseño, que son componentes críticos del QMSR.
- Capacitación: Comience a capacitar a su personal sobre las nuevas regulaciones para asegurar que todos los involucrados estén al tanto de los cambios y comprendan sus roles en el mantenimiento del cumplimiento.
- Integración: Comience a integrar los nuevos requisitos del QMSR en sus operaciones diarias para facilitar la transición.
Año 2:
- Continuar con la implementación de cambios en su sistema de calidad, asegurando que todas las actualizaciones estén completamente integradas y operativas.
- Realizar auditorías internas exhaustivas para verificar que las actualizaciones son efectivas y que su sistema de calidad está totalmente alineado con los requisitos del QMSR.
- Aborde rápidamente cualquier hallazgo de las auditorías internas para asegurar que todos los aspectos de su sistema de calidad cumplan con la normativa.
- Al final del segundo año, su sistema de calidad debería cumplir plenamente con el QMSR, y debería estar preparado para las inspecciones de la USFDA con la confianza de que no habrá problemas importantes.
Al seguir esta hoja de ruta, los fabricantes no solo pueden cumplir los requisitos de la USFDA, sino también establecer un sistema de calidad robusto que sea eficiente, estandarizado y reconocido a nivel mundial. Este enfoque proactivo ayudará a garantizar una transición fluida a las nuevas reglamentaciones y a mantener los más altos estándares de calidad y seguridad para los Dispositivos Médicos.
Conclusión: Adoptar medidas proactivas
Prepararse para las inspecciones de la USFDA bajo la nueva norma QMSR no se trata solo de evitar sanciones, sino de mejorar la seguridad y eficacia de los dispositivos. Al armonizar con la ISO 13485, la USFDA está estableciendo expectativas más altas, pero también proporcionando un camino hacia un cumplimiento global más simplificado. Los fabricantes que comiencen a adaptarse temprano deben centrarse en áreas clave como la gestión de riesgos y la vigilancia post-comercialización, y asegurarse de que sus sistemas de calidad estén actualizados y sean robustos, lo que no solo cumplirá con las expectativas reglamentarias sino que también mejorará su ventaja competitiva en el mercado.
