
5 min de lectura
El Programa de Auditoría Única de Dispositivos Médicos (MDSAP) permite que una Organización de Auditoría (AO) reconocida realice una única auditoría del Sistema de Gestión de Calidad (SGC) de un fabricante de Dispositivos Médicos. Proporciona requisitos reglamentarios relevantes para cinco países, es decir, Brasil (ANVISA), EE. UU. (FDA), Japón (PMDA), Canadá (Health Canada) y Australia (TGA). Además de las autoridades reglamentarias participantes, varios otros socios internacionales (los observadores oficiales y los miembros afiliados) están involucrados en el MDSAP.
La certificación MDSAP es obligatoria por parte de Health Canada para los dispositivos de Clase II, III y IV, pero es voluntaria para los otros cuatro países. Ha promovido la transparencia y la alineación reglamentaria entre las autoridades participantes y ha minimizado la necesidad de múltiples auditorías, ahorrando así tiempo y recursos a los fabricantes de Dispositivos Médicos. Para ofrecerle una mejor perspectiva sobre el programa MDSAP, aquí hemos intentado abordar las quince (15) preguntas más frecuentes.
- ¿Por qué se desarrolló el Programa MDSAP cuando ya existe una certificación ISO 13485 aceptada a nivel mundial?
MDSAP se desarrolló para reducir la carga de las auditorías reglamentarias para los fabricantes de Dispositivos Médicos y para promover una mayor alineación de los enfoques reglamentarios y los requisitos técnicos basados en estándares internacionales y mejores prácticas. Se centra en aportar coherencia, previsibilidad y transparencia a los programas reglamentarios mediante la estandarización de los procedimientos y prácticas de los reguladores y las organizaciones de auditoría de terceros.
La auditoría se basa en los requisitos del SGC según la ISO 13485 y los requisitos reglamentarios del país participante donde se comercializarán los Dispositivos Médicos.
- ¿Cuáles son los criterios de elegibilidad para someterse a una auditoría MDSAP?
Cualquier fabricante de Dispositivos Médicos que tenga la intención de comercializar su dispositivo en los países participantes puede someterse a una auditoría MDSAP. Sin embargo, cada Autoridad Reglamentaria puede establecer criterios de exclusión para ciertas condiciones si es necesario.
Por ejemplo, en Japón, las excepciones para la elegibilidad son:
- Un Sitio de Fabricación Registrado (RMS) que fabrica Dispositivos Médicos hechos de tejidos humanos/animales
- Un RMS que fabrica IVD radiactivos, y
- El establecimiento de un Titular de la Autorización de Comercialización (MAH)
- ¿La auditoría MDSAP incluye productos combinados?
Los Dispositivos Médicos que incluyen medicamentos (sustancias medicinales) o productos biológicos (por ejemplo, materiales de origen animal que han sido inactivados, o tejidos, células, o sustancias de origen microbiano o recombinante, sangre humana o extractos de sangre humana o productos sanguíneos, etc.) se consideran productos combinados y pueden incluirse en el alcance de una auditoría MDSAP.
Sin embargo, debido a las diferencias en cómo se regulan estos productos en las jurisdicciones de las Autoridades Reglamentarias participantes, los informes de auditoría y los documentos de certificación de MDSAP pueden no considerarse una alternativa a los requisitos de inspección y evaluación en algunas jurisdicciones.
Australia- Los productos combinados están sujetos a un examen externo de la TGA bajo la Evaluación de Conformidad Australiana. Pero una auditoría MDSAP efectiva puede reducir las inspecciones para estos dispositivos.
Brasil, Japón- Los productos combinados considerados Dispositivos Médicos están incluidos en el MDSAP, ya que no existen requisitos específicos en cuanto al QMS.
Canadá- El modelo MDSAP cubre los requisitos del QMS para los productos combinados considerados Dispositivos Médicos.
EE. UU.- Las auditorías MDSAP no se consideran alternativas a las inspecciones de la FDA para productos combinados.
- ¿Puedo seleccionar el país incluido en el alcance de la auditoría MDSAP?
Sí, la auditoría se realiza de acuerdo con el alcance declarado en la solicitud de servicios de certificación. Se espera que los fabricantes de Dispositivos Médicos cumplan con las regulaciones solo en las jurisdicciones donde se comercializarán sus productos.
- Soy un fabricante de Dispositivos Médicos de los US, con la intención de comercializar mi dispositivo solo en Japón. Estoy a punto de someterme a una auditoría MDSAP. ¿Necesito cumplir también con los requisitos de otros países?
No, solo se espera que los fabricantes de dispositivos médicos cumplan con los requisitos y regulaciones de ISO 13485 en las jurisdicciones donde se comercializarán sus productos.
- Mi Organización de Auditoría (AO) y mi Organismo Notificado Europeo son los mismos. ¿Puedo ser auditado para ambos al mismo tiempo?
Si su AO y el Organismo Notificado Europeo son el mismo, la evaluación de la conformidad puede realizarse después de llevar a cabo la auditoría MDSAP, no simultáneamente. Los Organismos Notificados Europeos son observadores de MDSAP, y la evaluación de la conformidad se realiza según el EU MDR 2017/745. Para MDSAP, la evaluación se lleva a cabo según los requisitos de ISO 13485 y los requisitos reglamentarios de los países participantes incluidos en el alcance.
- ¿Cuál es la diferencia entre las evaluaciones de Etapa I y II?
El proceso de auditoría inicial del MDSAP consta de dos etapas. La auditoría inicial, también denominada auditoría de Certificación Inicial, consta de auditorías de Etapa I y Etapa II.
La auditoría de la Etapa I incluye la revisión de la documentación y la evaluación de la preparación del fabricante de dispositivos médicos para someterse a una auditoría de la Etapa II.
La auditoría de la Etapa II se realiza para verificar si todos los requisitos aplicables de ISO 13485 y otros requisitos reglamentarios de la autoridad reglamentaria correspondiente están implementados.
- ¿Cuántos auditores puedo esperar para una auditoría MDSAP?
La Determinación del Tiempo de Auditoría especifica cómo determinar la duración de la auditoría in situ en días-hombre. El AO decide cuántos auditores compondrán el equipo de auditoría. Por ejemplo, una auditoría de (06) días-hombre puede completarse en tres (03) días por un equipo de dos (02) auditores.
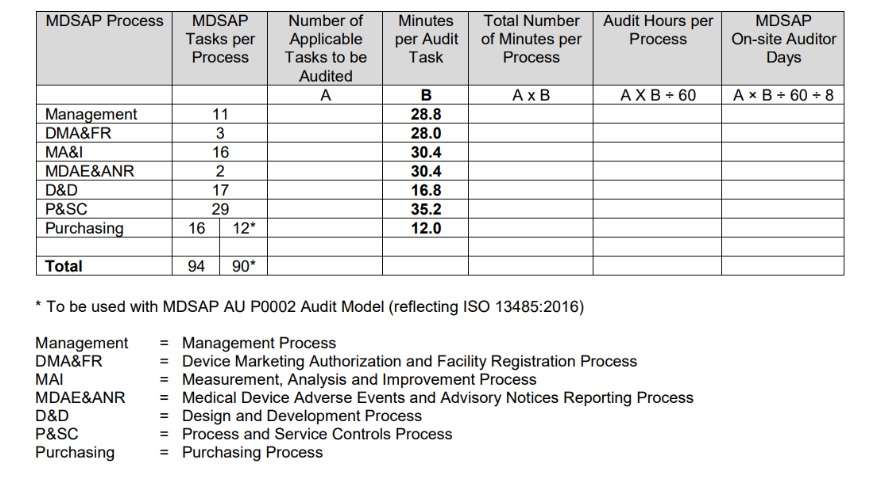
- ¿Cómo se programa la auditoría MDSAP?
El Procedimiento de Determinación del Tiempo de Auditoría, emitido por la FDA, resume el proceso para determinar la duración del cálculo de la auditoría en la siguiente tabla.
El cálculo de la duración de la auditoría se basa principalmente en el número de tareas de auditoría aplicables asociadas con el tipo de auditoría a realizar y las actividades específicas de la organización a auditar.
Para obtener información detallada al respecto, puede consultar MDSAP P0008007
- ¿Existe una guía o una lista de verificación a la que pueda acceder para asegurar el cumplimiento con una auditoría MDSAP?
Sí, puede acceder al documento MDSAP Audit Approach. Es una guía bien organizada emitida por la USFDA que hace referencia cruzada a secciones específicas de la ISO 13485:2016 y a las regulaciones relevantes emitidas por la TGA de Australia, la ANVISA de Brasil, Health Canada de Canadá, el MHLW/PMDA de Japón y la US FDA.
- ¿Cuál es la función de un observador en una auditoría MDSAP?
Un observador MDSAP es una Autoridad Reglamentaria a la que se le permite asistir a reuniones, evaluaciones y otras actividades, pero que no utiliza los entregables de MDSAP. Los observadores están representados en el Consejo de Autoridades Reglamentarias de MDSAP (RAC) por un gerente de alto nivel.
- ¿Cuáles son los próximos pasos a seguir si he recibido una puntuación de 4 o superior?
El sistema de calificación se aplica a las no conformidades observadas durante la auditoría por parte de la AO. Una puntuación de 4 o 5 indica un alto riesgo de intervención. Debe proporcionar un plan de subsanación para cada no conformidad registrada en un plazo de 15 días naturales a partir de la fecha de emisión del informe de no conformidad. El plan de subsanación debe incluir los resultados de la investigación de la no conformidad, sus causas y las acciones correctivas planificadas para evitar cualquier recurrencia. La evidencia de la implementación del plan/acción de subsanación debe proporcionarse dentro de los treinta (30) días naturales posteriores a la fecha de finalización de la auditoría.
- ¿Existe una diferencia en el proceso de abordar la auditoría por un auditor interno frente a un AO?
MDSAP sigue un enfoque basado en procesos. Es probable que el Organismo Auditor (AO) examine las conexiones y los hilos, mientras que un auditor interno podría centrarse más en un aspecto funcional a la vez. Por lo tanto, el Organismo Auditor (AO) podría encontrar una no conformidad en un área funcional y buscar respuestas en un área funcional diferente. Sin embargo, seguir el enfoque basado en procesos podría ser disruptivo durante una auditoría interna.
- ¿Puedo apelar ante la AO si puedo demostrar que una no conformidad registrada no es válida?
AO tiene un proceso de apelación o disputa, que puede utilizar si puede demostrar que una no conformidad registrada no es válida. Sin embargo, las calificaciones asignadas a las no conformidades no pueden modificarse debido a acciones correctivas. Solo pueden modificarse basándose en pruebas que demuestren que no eran válidas.
- ¿Cuánto tiempo es válido el certificado MDSAP?
Los fabricantes de Dispositivos Médicos certificados bajo el programa MDSAP serán auditados anualmente, de acuerdo con un ciclo de certificación de tres años. La auditoría inicial es una auditoría completa del QMS del fabricante de Dispositivos Médicos. Le siguen auditorías de vigilancia realizadas anualmente durante dos (02) años consecutivos. El ciclo se reinicia con una auditoría de recertificación en el tercer año.
Para saber más sobre nuestros servicios de MDSAP, Contacte a Freyr hoy mismo para programar una llamada con nuestros Expertos.









