4 min de lectura
La Notificación de Dispositivos Médicos (MDR) es una herramienta de vigilancia post-comercialización que la FDA (Administración de Alimentos y Medicamentos) utiliza para supervisar el rendimiento de los dispositivos, detectar posibles problemas de seguridad relacionados con ellos y contribuir a las evaluaciones de riesgo-beneficio de los dispositivos. El propósito de la MDR es detectar y abordar los eventos adversos relacionados con los dispositivos de manera oportuna. Permite a médicos, centros de salud, fabricantes y consumidores realizar notificaciones voluntarias para comprender la seguridad y eficacia del dispositivo tras su comercialización.
El MDR es aplicable a todas las clases de Dispositivos Médicos que se fabrican en los Estados Unidos de América (EE. UU.) o se importan a los EE. UU. Los fabricantes de Dispositivos Médicos que deseen comercializar sus dispositivos en los EE. UU. deben cumplir con el MDR, ya que, de lo contrario, podrían enfrentar sanciones económicas. Es aplicable en los EE. UU., incluyendo un evento extranjero, es decir, es aplicable a los Dispositivos Médicos comercializados legalmente en los Estados Unidos, tanto los fabricados en los EE. UU. como en países extranjeros. Además, existen varios casos de aplicabilidad para un MDR, como:
- si un dispositivo se fabrica en los EE. UU., se distribuye localmente y a otros mercados
- cuando un dispositivo se fabrica en US pero se distribuye en otros mercados
- cuando un dispositivo se fabrica en el país extranjero, se suministra en los EE. UU. y otros mercados
- cuando un dispositivo se fabrica en el país extranjero y se distribuye localmente y
- cuando un dispositivo está bajo investigación en US
MDR y el flujo del proceso de informes
El reglamento MDR contiene muchos requisitos obligatorios para que los fabricantes, importadores e instalaciones de usuarios de dispositivos informen a la FDA sobre ciertos eventos adversos relacionados con dispositivos y problemas de productos. El diagrama de flujo del proceso que se presenta a continuación detalla el proceso de notificación paso a paso.
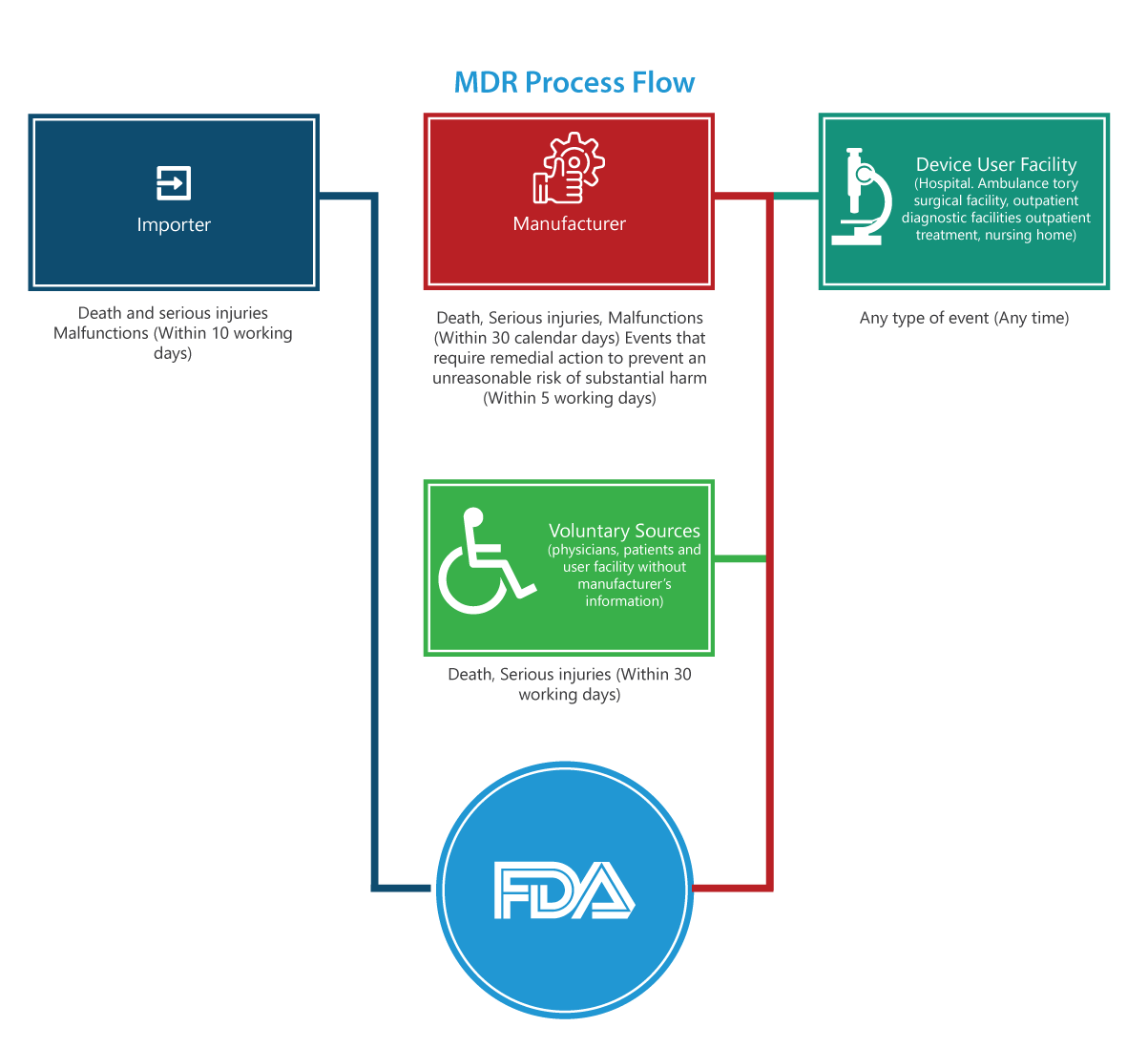
¿A quién se aplica?
Importadores
Los informes de muertes, lesiones graves y mal funcionamiento deben presentarse a la FDA y al fabricante en un plazo de 30 días hábiles. Si el mal funcionamiento puede causar lesiones o muertes en otros lugares, los importadores deben informar del mal funcionamiento al fabricante.
Fabricantes
Los informes de un evento (muertes, lesiones graves y mal funcionamiento) designado por la FDA o un evento que requiera una acción correctiva para prevenir un riesgo irrazonable de daño sustancial a la salud pública deben presentarse a la FDA en un plazo de 5 días hábiles completando el formulario 3500A.
Instalación de usuario de Dispositivos Médicos (Hospital, centro quirúrgico ambulatorio, residencia de ancianos, centro de diagnóstico ambulatorio o centro de tratamiento ambulatorio)
Los informes deben presentarse al fabricante del dispositivo a más tardar 10 días hábiles después del día en que tenga conocimiento de información de que un dispositivo ha causado o puede haber contribuido a una lesión grave a un paciente de la instalación. Si el fabricante es desconocido, la instalación debe presentar el informe a la FDA.
Grupos voluntarios
Los pacientes, profesionales de la salud y consumidores que encuentren un problema relacionado con un dispositivo médico pueden informarlo a la FDA a través de MedWatch
eMDR
La FDA hizo obligatorio el MDR electrónico (eMDR) en 2015 para identificar problemas críticos de calidad e integridad de los datos asociados con la notificación de lesiones graves relacionadas con todas las clases de Dispositivos Médicos. Se prefiere que el eMDR sea el modo de notificación.
Los fabricantes pueden presentar su eMDR a través de una Pasarela de Envíos Electrónicos (ESG). Desde el momento de la presentación, la pasarela electrónica tarda hasta 48 horas en enviar un acuse de recibo. Si hay algún error al presentar el informe, aparecerá un mensaje para realizar la(s) corrección(es).
eMDR – ¿Cómo es beneficioso?
eMDR ofrece múltiples ventajas sobre el mecanismo de notificación manual (es decir, MDR). A continuación se enumeran algunos beneficios notables en los que los fabricantes / agencias / pacientes pueden confiar:
- La herramienta de envío de eMDR mejora la colaboración entre una organización, la agencia de salud (FDA) y los pacientes.
- eMDR ahorra costes. La automatización reduce la necesidad de gastos administrativos y comunicación tradicional; ayuda a acelerar el proceso y fomenta la notificación eficaz de eventos, lo que resulta en una interacción inmediata con la FDA.
- Los procesos manuales implican una cantidad considerable de papeleo, pueden ser largos y difíciles de rastrear y procesar. La presentación de eMDR está automatizada y centralizada. Los registros se pueden recuperar fácilmente, ahorrando mucho tiempo durante la revisión.
- eMDR permite a las partes señalar rápidamente los errores de envío, a diferencia de las correspondencias manuales y que consumen mucho tiempo con la FDA.
- eMDR actúa como un único punto de entrada para procesar todas las presentaciones electrónicas en un entorno altamente seguro y es beneficioso porque las quejas en la organización pueden vincularse directamente al formulario MedWatch e integrarse dentro de la pasarela de la FDA.
eMDR y el flujo del proceso de informes
El reglamento eMDR contiene requisitos obligatorios para que los fabricantes, importadores e instalaciones de usuarios de dispositivos informen a la FDA sobre ciertos eventos adversos relacionados con dispositivos y problemas de productos. El diagrama de flujo a continuación detalla el proceso de notificación paso a paso.
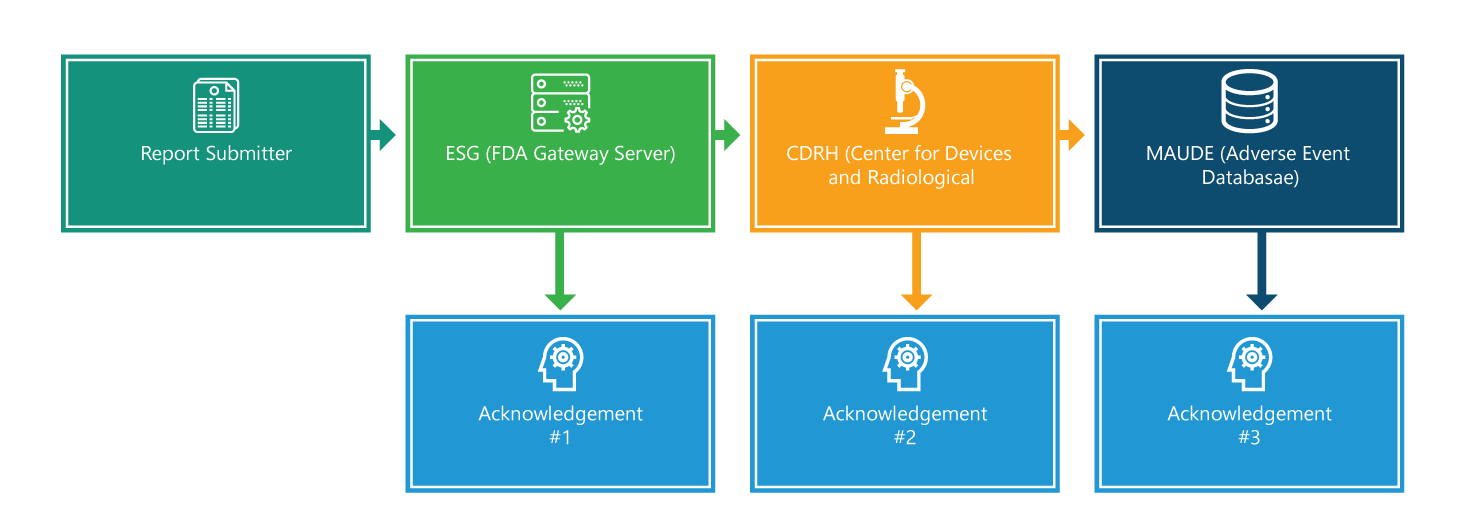
El proceso de notificación consta de cuatro pasos. Excepto el primero, cada paso es confirmado. Además, cada paso cuenta con información adicional que ayudará a facilitar el proceso.
Paso 1: Remitente del informe
Presentación de un eMDR. Al principio, para realizar una presentación, se debe tener una firma electrónica y se debe asegurar que los nombres de archivo de la presentación incluyan solo un punto, que se utiliza para indicar la extensión del tipo de archivo (por ejemplo, 555.xml o 555.pdf). Sin embargo, el tiempo de entrega y procesamiento de la solicitud depende del tamaño total de su presentación; las presentaciones más grandes tardan más en ser entregadas y procesadas.
Paso 2: Pasarela de Envíos Electrónicos (ESG)
Cuando su presentación llegue al ESG, debería recibir rápidamente un acuse de recibo #1, a menos que el ESG esté fuera de servicio por mantenimiento. Debe verificar el estado de su MDR en el sitio web del ESG.
Paso 3: CRDH
eMDR se envía automáticamente desde el ESG al Centro de Dispositivos y Salud Radiológica (CDRH). Una vez enviado, como en el paso 2, debería recibir una confirmación, es decir, el n.º 2.
Paso 4: Experiencia de Dispositivos de Fabricantes y Usuarios (MAUDE)
Cuando el CDRH valida y actualiza la presentación en la base de datos de Eventos Adversos (MAUDE), se espera que el remitente reciba un acuse de recibo #3. Cabe señalar que cualquier error que ocurra durante la validación y carga se registra.
La Notificación de Dispositivos Médicos (MDR) es un proceso fundamental que ayuda a salvar vidas y a proteger a los pacientes de riesgos innecesarios. Garantiza que todas las partes implicadas en la atención al paciente sean responsables y estén atentas al usar los dispositivos.
eMDR facilita la presentación de informes, pero la documentación y el seguimiento pueden consumir muchos recursos. Hágalo bien a la primera; hable con nosotros en sales@freyrsolutions.com.