2 min de lectura
El objetivo principal de la US FDA es examinar constantemente y reducir la brecha entre los procesos reglamentarios para la importación y venta ininterrumpida de Dispositivos Médicos nuevos y de alta calidad en el mercado de US. En 1998, la US FDA lanzó un programa llamado «El nuevo paradigma 510(k): enfoques alternativos para demostrar la equivalencia sustancial en las notificaciones previas a la comercialización». Su objetivo es establecer una vía eficiente de presentación 510(k) de la FDA que contenga ciertos cambios en la solicitud 510(k) ya aprobada. Esta nueva notificación 510(k) ofrece (03) tres tipos de presentaciones: 510(k) especial, 510(k) abreviada y 510(k) tradicional. En 2019, la US FDA publicó un documento de orientación especial 510(k) que describe una vía opcional para los fabricantes que realizan ciertas modificaciones bien definidas a su dispositivo comercializado legalmente.
¿Por qué una 510(k) especial?
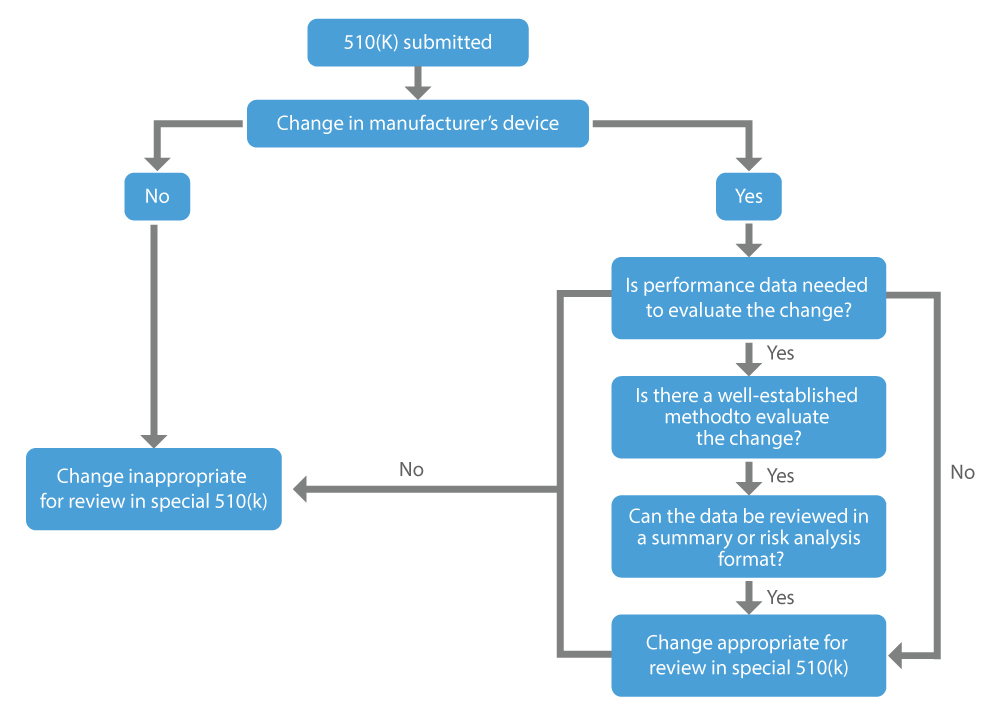
Cuando un fabricante desea la aprobación de las modificaciones que ha realizado en un dispositivo ya comercializado, es decir, el dispositivo existente, puede solicitar una 510(k) especial. Los principales factores a considerar al determinar si un cambio en un dispositivo existente puede ser apropiado para una 510(k) especial son los siguientes:
- El cambio se realiza en el propio dispositivo precedente legalmente comercializado del solicitante.
- No se requieren datos de rendimiento, o se dispone de métodos bien establecidos si se considera necesario para evaluar el cambio.
- Todos los datos de rendimiento para respaldar una determinación de equivalencia sustancial pueden revisarse en formato de resumen o análisis de riesgos.
Documentos requeridos para el 510(k) especial.
- Carta de presentación
- El nombre del dispositivo (existente) legalmente comercializado por el fabricante y el número de 510(k).
- Una descripción detallada de los cambios realizados en el dispositivo que resultaron en la presentación de un nuevo 510(k)
- Una comparación del dispositivo modificado con el dispositivo autorizado en formato tabular
- Otros cambios en el etiquetado o el diseño.
- Un resumen conciso de las actividades de control de diseño
- Según el análisis de riesgos, una identificación de las actividades de verificación y/o validación necesarias para cumplir con 21 CFR 820.30
- Formulario de indicaciones de uso
- Una declaración de que el solicitante ha cumplido y no está actualmente en violación de los requisitos del procedimiento de control de diseño según lo especificado en 21 CFR 820.30 y que los registros están disponibles para su revisión previa solicitud
Plazo de Revisión Especial 510(k) por la FDA de US
Según las directrices de la FDA «Política de rechazo de aceptación para 510(k)», el plazo de revisión para las presentaciones especiales de 510(k) es de treinta (30) días a partir de su recepción.
¿Cuándo solicitar una 510(k) especial?
La US FDA realiza esfuerzos constantes para proporcionar Dispositivos Médicos seguros y eficaces para promover la salud humana. El programa especial 510(k) es eficiente y coherente con el procedimiento de revisión menos oneroso que ayuda a los fabricantes extranjeros a vender sus dispositivos dentro de los EE. UU. y permite a los pacientes acceder a tiempo a nuevos Dispositivos Médicos.
Para cualquier aclaración adicional sobre el proceso especial 510(k) de la FDA, contacte con Freyr, un experto reglamentario probado. Manténgase informado. Manténgase en cumplimiento.