

3 min di lettura
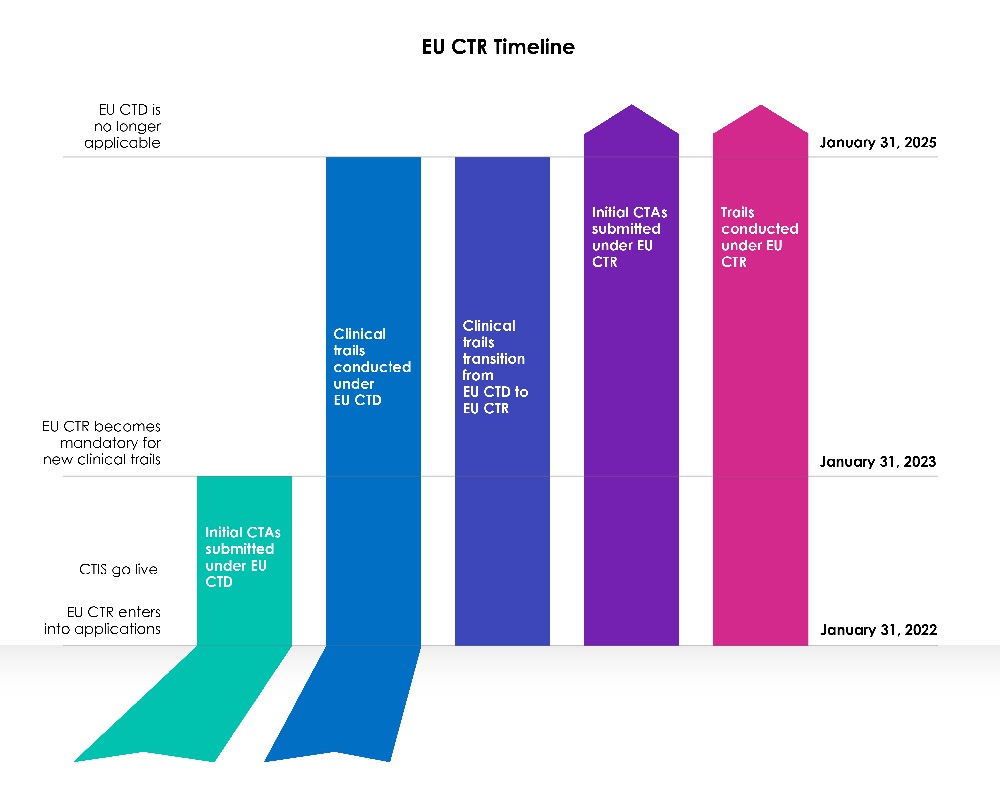
A partire dal 31 gennaio 2022, il nuovo regolamento farmaceutico dell'Unione Europea (UE) per le sperimentazioni cliniche (Regolamento sulle Sperimentazioni Cliniche - CTR) è diventato obbligatorio, abrogando la Direttiva sulle Sperimentazioni Cliniche 2001/20/CE. Il regolamento armonizza i protocolli per la valutazione e la supervisione delle sperimentazioni cliniche in tutta l'UE. Le linee guida sono state riviste per promuovere un approccio uniforme alla ricerca clinica, ponendo l'accento sulla sicurezza dei partecipanti alle sperimentazioni cliniche e su una maggiore divulgazione pubblica.
Il regolamento stabilisce un nuovo sistema di valutazione in due parti per tutti gli studi clinici nell'UE. La Parte I consiste in una valutazione scientifica della documentazione principale dello studio clinico, mentre la Parte II prevede una valutazione etica della documentazione a livello nazionale. A seguito di questa valutazione in due parti, ogni Stato membro prenderà una decisione unificata sullo studio e notificherà lo sponsor tramite il Sistema Informativo sugli Studi Clinici.
Tempistiche di Transizione per i Nuovi Richiedenti
Una fase di transizione di tre anni è iniziata con la data di attivazione del CTIS dell'UE.
Anno 1 (31 gennaio 2022 - 30 gennaio 2023):
La Direttiva dell'Unione Europea (UE) sulle Sperimentazioni Cliniche 2001/20/CE (EU-CTD) ha regolamentato le sperimentazioni cliniche nell'UE dal 2004. Si è sforzata di standardizzare le regole e ha migliorato significativamente la sicurezza dei pazienti nelle sperimentazioni cliniche. Tuttavia, in pratica, ha creato conseguenze indesiderate. Durante il primo anno, a seguito dell'implementazione del CTIS, agli sponsor è stato permesso di scegliere se presentare una nuova Domanda di Sperimentazione Clinica (CTA) nell'ambito del Clinical Trial Information System (CTIS) ai sensi della Direttiva sulle Sperimentazioni Cliniche (CTD: Direttiva 2001/20/CE), o di utilizzare il CTIS in conformità con la legislazione attuale, il Regolamento sulle Sperimentazioni Cliniche (UE) n. 536/2014.
Entrambe le idee erano valide e gli sponsor hanno avuto la possibilità di scegliere quale legislazione seguire.
I membri erano pronti a utilizzare il Clinical Trial Information System (CTIS) e hanno accettato le domande ai sensi della nuova legislazione, il Regolamento sulle Sperimentazioni Cliniche (EU CTR), il primo giorno di operatività del CTIS.
Anni 2 e 3 (31 gennaio 2023 - 31 gennaio 2025):
A partire dal 31 gennaio 2023, tutte le nuove domande CT devono essere presentate tramite il CTIS ai sensi della nuova legislazione (CTR).
Le nuove domande di CT non possono essere presentate in EudraCT ai sensi della Direttiva sulle Sperimentazioni Cliniche (CTD). La Direttiva UE sulle Sperimentazioni Cliniche non consente più l'ammissione di nuovi Member States dopo il 31 gennaio 2023. Le sperimentazioni condotte ai sensi della CTD devono prima essere transitate, dopodiché una domanda aggiuntiva relativa a un Member State può essere presentata tramite EU CTIS.
Per i richiedenti esistenti
Le domande di sperimentazione clinica (CT) presentate prima del 30 gennaio 2023, ai sensi della vecchia legislazione (CTD) e utilizzando EudraCT, saranno autorizzate a proseguire fino al completamento ai sensi di tale Direttiva ((CTD: Direttiva 2001/20/CE), fino al 30 gennaio 2025. Le procedure rimarranno invariate e gli sponsor potranno presentare modifiche significative e notifiche di fine sperimentazione come richiesto dal regolamento. EudraCT rimarrà attivo durante il periodo di transizione per consentire la continuazione di queste sperimentazioni.
È tuttavia importante notare che le domande di transizione possono essere presentate in qualsiasi momento durante il periodo di transizione di tre anni, e si incoraggiano gli sponsor a completare il processo con sufficiente anticipo nel periodo di transizione per garantire la continuità delle sperimentazioni cliniche nell'UE oltre il 30 gennaio 2025, considerando le festività legali e la sospensione invernale di due settimane.
Studi non trasferibili
- Gli studi clinici che si sono conclusi o si concluderanno poco prima della fine del periodo di transizione EU/EEA non devono essere trasferiti.
- Se una notifica di fine studio è stata completata in tutti i paesi membri dell'UE/EEA, ma la fine globale dello studio non è ancora stata notificata, lo studio non dovrebbe essere trasferito. Ai sensi della Direttiva, la fine globale dello studio e i risultati riassuntivi dello studio dovrebbero essere pubblicati tramite EudraCT.
- Gli studi iniziati prima dell'attuazione della Direttiva 2001/20/CE non beneficiano di tale procedura di transizione. Se sono interventistici e devono continuare a operare dopo la fine della fase di transizione del CTR, deve essere presentata una nuova CT Application ai sensi del CTR.
- Anche gli studi pediatrici condotti al di fuori dell'UE/EEA, ma per i quali è stato assegnato un numero EudraCT, non devono essere convertiti.
- Gli studi sospesi dopo la fine del periodo di transizione non possono essere oggetto di transizione. Il riavvio dello studio in queste circostanze richiederebbe la presentazione di una nuova CT Application ai sensi del CTR.
Il CTIS dell'UE riceve regolarmente aggiornamenti tecnici dall'EMA per migliorarne le caratteristiche e le funzionalità. Quando vengono apportate modifiche importanti al CTIS, l'EMA pubblica note di rilascio che descrivono ciò che è cambiato nel sistema. Gli aggiornamenti possono includere miglioramenti alle caratteristiche e funzionalità esistenti, l'aggiunta di nuove funzionalità e miglioramenti funzionali e tecnici. Un partner normativo esperto può affrontare le potenziali sfide e aiutare gli sponsor nella transizione degli studi clinici esistenti e futuri, nell'ambito delle strategie di sviluppo clinico. Clicca qui per saperne di più sul CTIS e sull'esperienza di Freyr in questo settore: https://www.freyrsolutions.com/medical-devices/regulatory-affairs.









