
Panoramica dei servizi di conformità al regolamento IVDR dell'UE
Il regolamento europeo IVDR (In Vitro Diagnostic Regulation) costituisce una nuova base normativa per l'immissione sul mercato europeo dei dispositivi medici per la diagnostica in vitro. Il regolamento IVDR dell'UE è destinato a sostituire l'attuale direttiva UE sui dispositivi medici diagnostici in vitro (UE 98/79/CE). In quanto regolamento europeo, sarà applicabile in tutti gli Stati membri dell'UE e negli Stati membri dell'Associazione europea di libero scambio (AELS), senza che sia necessario recepirlo nel diritto degli Stati interessati. In qualità di partner normativo affermato, Freyr offre alle aziende servizi di conformità e consulenza in materia di IVDR all'interno dell'UE, al fine di garantire la conformità alle normative sulla diagnostica in vitro e alla marcatura CE.
Fissate un appuntamento con i nostri esperti UE in materia di IVDR
Il regolamento IVDR - Calendario di transizione
Prevista per entrare in vigore il 26 maggio 2022, la nuova normativa europea IVDR è considerata un cambiamento significativo nella regolamentazione dei dispositivi medici per la diagnostica in vitro. Ecco una panoramica della transizione verso l'IVDR:
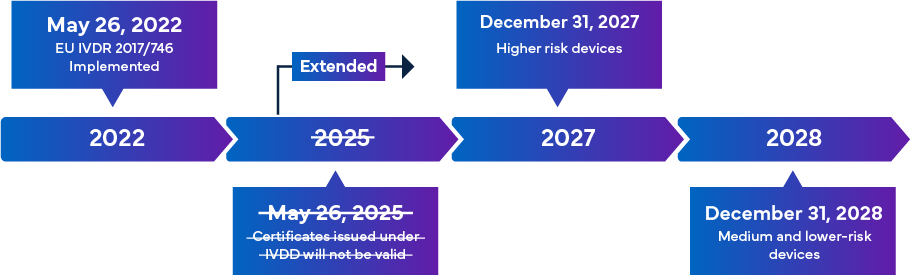
Una volta pienamente attuate, le norme di conformità dell'IVDR garantiscono che quasi tutti i dispositivi diagnostici in vitro (IVD) immessi sul mercato europeo siano sottoposti all'esame dell'organismo notificato (ON) dell'IVDR e alla certificazione del marchio CE nell'ambito della procedura di approvazione degli IVD. In questo scenario, oltre all'allineamento alla classificazione aggiornata dell'IVDR, i fabbricanti devono rivedere immediatamente la documentazione tecnica chiave per ottenere la registrazione dell'IVD e il marchio CE. I requisiti dell'IVDR variano a seconda della classe di rischio dell'IVD. In generale, per la certificazione degli IVD, i fabbricanti devono effettuare le seguenti operazioni:
- Esame dei fascicoli tecnici in conformità al regolamento IVDR (Regolamento UE 2017/746)
- Preparazione del rapporto di valutazione delle prestazioni (PER) per tutti i dispositivi diagnostici in vitro (IVD)
- Monitoraggio delle prestazioni post-commercializzazione (PMPF) ai sensi dell'allegato XIII, parte B, dell'IVDR.
- Relazione di vigilanza ai sensi dell'articolo 82 dell'IVDR
Freyr supporta i propri clienti nella realizzazione di una revisione sistematica della letteratura scientifica e li aiuta a pianificare e preparare un PER (Piano di valutazione del rischio) per conformarsi alla normativa sui dispositivi diagnostici in vitro. Freyr dispone di esperti specializzati che forniscono servizi di consulenza in materia di IVDR e supporto alla sorveglianza post-commercializzazione (PMS), che è parte integrante del sistema di gestione della qualità del fabbricante, nonché al monitoraggio delle prestazioni post-commercializzazione (PPM).
Ricevi consigli da esperti sulla conformità al regolamento IVDR dell'UE
Servizi di conformità al regolamento IVDR dell'UE: COMPETENZA E VANTAGGI
- Piano di transizione per la conformità al regolamento IVDR
- Esame tecnico e analisi delle lacune dei requisiti dell'IVDR relativi ai GSPR (requisiti generali di sicurezza e prestazione)
- Fornire assistenza nella compilazione del fascicolo tecnico in conformità con i requisiti dell'IVDR
- Relazioni sulla validità scientifica basate sulla letteratura e/o su dati interni
- Relazioni sulle prestazioni cliniche basate sulla letteratura e/o su dati interni
- Dati clinici o rapporti di valutazione delle prestazioni
- Protocolli e relazioni sul monitoraggio delle prestazioni post-commercializzazione (SPPM)
- Protocolli e relazioni di sorveglianza post-commercializzazione (SPM)
- Redigere/rivedere altri documenti quali foglietti illustrativi/IFU (istruzioni per l'uso), guide rapide (QRI), manuali d'uso/di funzionamento, ecc.

- Garantire la conformità al regolamento IVDR, la registrazione IVD e la marcatura CE
- Ottima conoscenza della normativa e competenza nei settori chiave interessati dall'IVDR dell'UE
- Modello di consegna incentrato sulla gestione del progetto per garantire il rispetto delle scadenze.
- Esperti interni dell'ON (esame della relazione da parte dei revisori interattivi dell'ON)
- Team specializzati con competenze trasversali in settori di impatto e categorie di dispositivi specifici
- Contributi trasversali di esperti nel settore dei dispositivi medici per garantire la conformità ai requisiti
- Insieme dei servizi relativi alla conformità, alla revisione e alla pianificazione
- Spiccata competenza nel garantire la coerenza dei risultati finali (tempistiche e qualità)
