4 minuti di lettura
La Segnalazione di Dispositivi Medici (MDR) è uno strumento di sorveglianza post-commercializzazione che la Food and Drug Administration (FDA) utilizza per monitorare le prestazioni dei dispositivi, rilevare potenziali problemi di sicurezza legati ai dispositivi e contribuire alle valutazioni del rapporto rischio-beneficio dei dispositivi. Lo scopo dell'MDR è rilevare e affrontare tempestivamente gli eventi avversi correlati ai dispositivi. Consente a medici, strutture sanitarie, produttori e consumatori di segnalare volontariamente per comprendere la sicurezza e l'efficacia del dispositivo dopo la commercializzazione.
L'MDR è applicabile a tutte le classi di dispositivi medici, che sono prodotti negli Stati Uniti d'America (USA) o importati negli USA. I produttori di dispositivi medici che intendono commercializzare i loro dispositivi negli USA devono conformarsi all'MDR, altrimenti ciò potrebbe comportare sanzioni finanziarie. È applicabile negli USA, inclusi eventi esteri, ovvero è applicabile ai dispositivi medici legalmente commercializzati negli Stati Uniti sia prodotti negli USA che in paesi esteri. Inoltre, ci sono vari casi di applicabilità per un MDR, come:
- se un dispositivo è fabbricato negli STATI UNITI D'AMERICA, distribuito localmente e in altri mercati
- quando un dispositivo è fabbricato negli STATI UNITI D'AMERICA ma distribuito in altri mercati
- quando un dispositivo è fabbricato nel paese estero, fornito negli STATI UNITI D'AMERICA e in altri mercati
- quando un dispositivo è fabbricato nel paese estero e distribuito localmente e
- quando un dispositivo è sotto indagine negli STATI UNITI D'AMERICA
MDR e il flusso del processo di segnalazione
Il regolamento MDR contiene molti requisiti obbligatori per produttori, importatori e strutture che utilizzano dispositivi, per segnalare alla FDA determinati eventi avversi correlati ai dispositivi e problemi del prodotto. Il diagramma di flusso del processo fornito di seguito illustra il processo di segnalazione passo dopo passo.
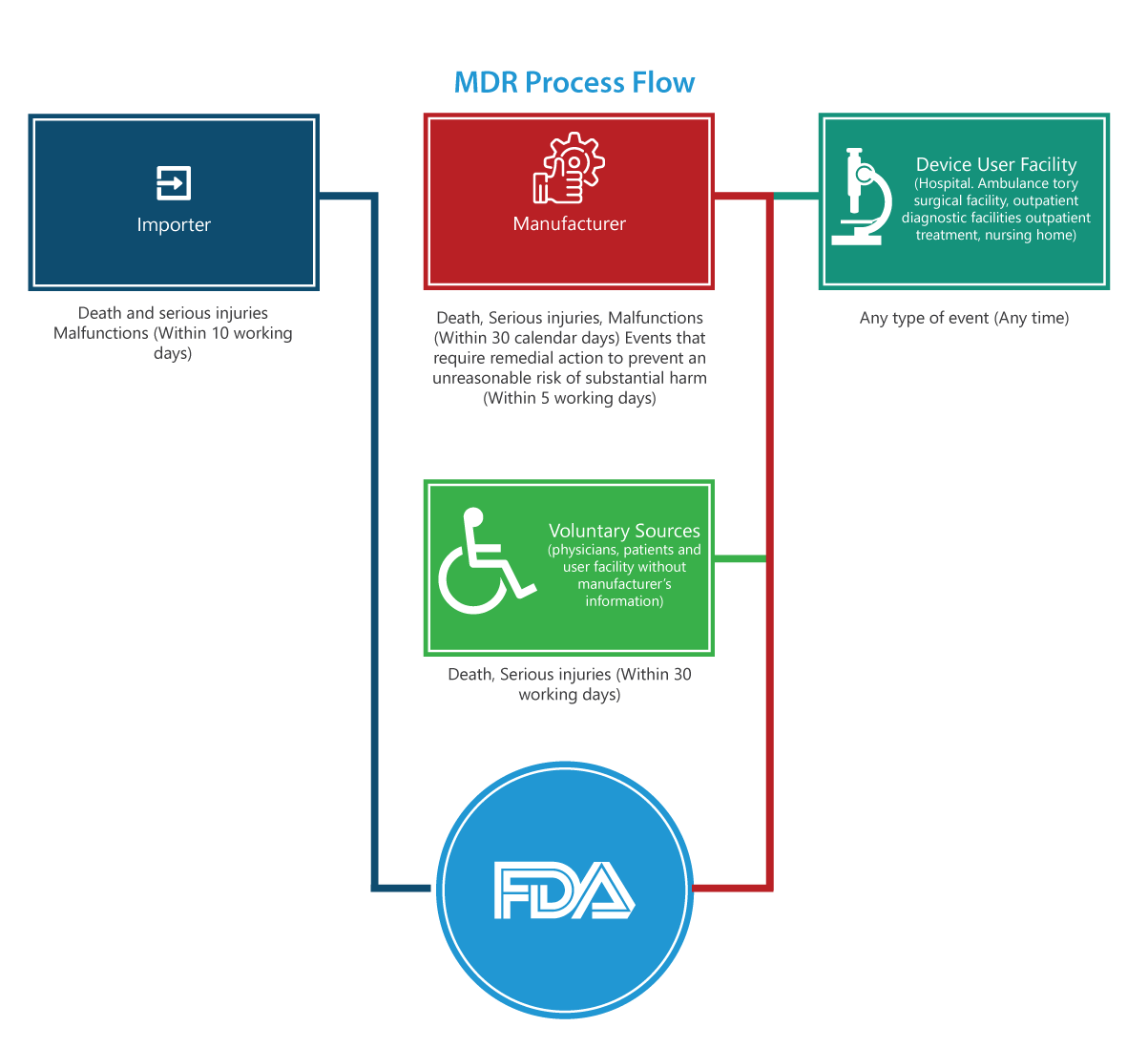
A chi si applica?
Importatori
Le segnalazioni di decessi, lesioni gravi e malfunzionamenti devono essere presentate alla FDA e al fabbricante entro trenta giorni lavorativi. Se il malfunzionamento può causare lesioni o decessi altrove, gli importatori devono segnalare il malfunzionamento al fabbricante.
produttori
Le segnalazioni relative a un evento (decessi, lesioni gravi e malfunzionamenti) designato dalla FDA o a un evento che richiede un'azione correttiva per prevenire un rischio irragionevole di danno sostanziale alla salute pubblica devono essere presentate alla FDA entro cinque giorni lavorativi compilando il modulo 3500A.
Struttura Sanitaria Utente (Ospedale, struttura chirurgica ambulatoriale, casa di cura, struttura diagnostica ambulatoriale o struttura di trattamento ambulatoriale)
Le segnalazioni devono essere presentate al fabbricante del dispositivo entro dieci giorni lavorativi dal giorno in cui viene a conoscenza di informazioni secondo cui un dispositivo ha causato o potrebbe aver contribuito a una lesione grave a un paziente della struttura. Se il fabbricante è sconosciuto, la struttura deve presentare la segnalazione alla FDA.
Gruppi volontari
Pazienti, professionisti sanitari e consumatori che riscontrano un problema relativo a un dispositivo medico possono segnalarlo alla FDA tramite MedWatch.
eMDR
La FDA ha reso obbligatorio l'MDR elettronico (eMDR) nel 2015 per identificare problemi critici di qualità e integrità dei dati associati alla segnalazione di lesioni gravi relative a tutte le classi di dispositivi medici. L'eMDR è la modalità di segnalazione preferita.
I produttori possono inviare il loro eMDR tramite un Electronic Submissions Gateway (ESG). Dal momento dell'invio, il gateway elettronico impiega fino a 48 ore per inviare una conferma. Se si verifica un errore durante l'invio del rapporto, verrà visualizzato un 'messaggio' per apportare le correzioni.
eMDR – Quali sono i suoi vantaggi?
L'eMDR offre numerosi vantaggi rispetto al meccanismo di segnalazione manuale (cioè, MDR). Di seguito sono elencati alcuni notevoli vantaggi su cui i produttori / l'agenzia / i pazienti possono contare:
- Lo strumento di invio eMDR migliora la collaborazione tra un'organizzazione, l'agenzia sanitaria (FDA) e i pazienti.
- L'eMDR riduce i costi. L'automazione diminuisce la necessità di oneri amministrativi e comunicazioni tradizionali; aiuta ad accelerare il processo e favorisce una segnalazione efficace degli eventi, consentendo un'interazione immediata con la FDA.
- I processi manuali comportano una notevole documentazione, possono essere lunghi e difficili da tracciare e gestire. L'invio eMDR è automatizzato e centralizzato. I registri possono essere recuperati facilmente, risparmiando molto tempo durante la revisione.
- eMDR consente alle parti di segnalare rapidamente gli errori di presentazione, a differenza delle corrispondenze manuali e dispendiose in termini di tempo con la FDA.
- eMDR funge da unico punto di accesso per elaborare tutte le presentazioni elettroniche in un ambiente altamente sicuro ed è vantaggioso perché i reclami presso l'organizzazione possono essere collegati direttamente al modulo MedWatch e integrati nel gateway della FDA.
eMDR e il flusso del processo di segnalazione
Il regolamento eMDR contiene requisiti obbligatori per produttori, importatori e strutture utilizzatrici di dispositivi di segnalare alla FDA determinati eventi avversi correlati ai dispositivi e problemi di prodotto. Il diagramma di flusso seguente descrive in dettaglio il processo di segnalazione passo dopo passo.
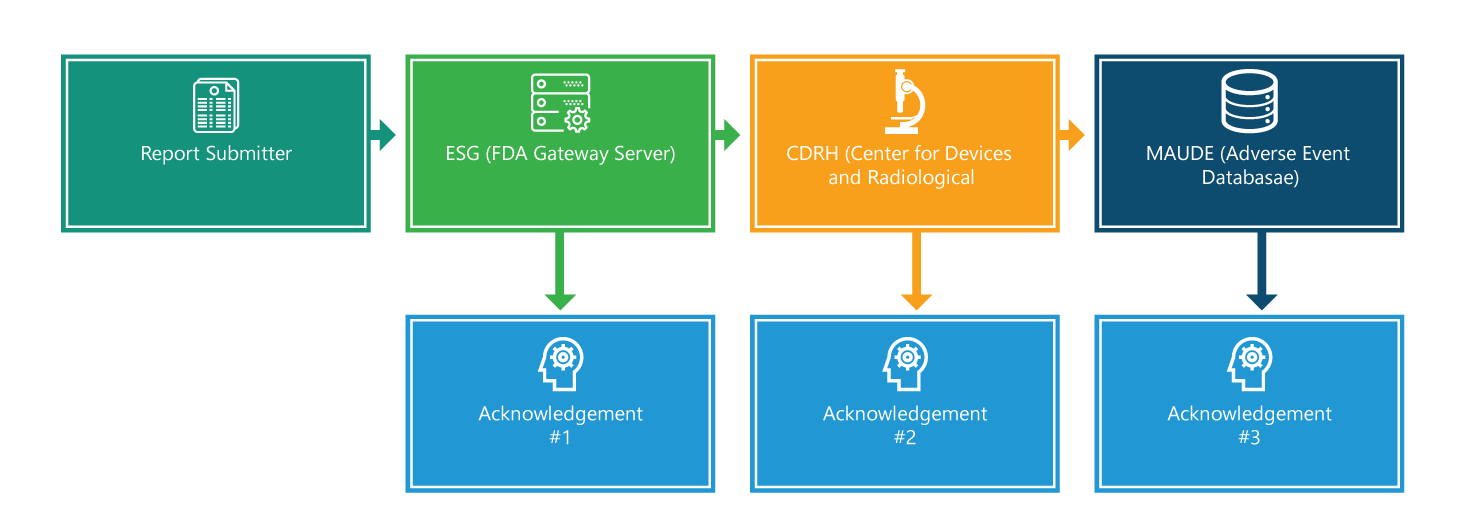
Il processo di segnalazione comprende quattro fasi. Ad eccezione della prima fase, ogni fase viene riconosciuta. Inoltre, ogni fase è corredata di informazioni aggiuntive che faciliteranno il processo.
Fase 1: Soggetto che presenta la segnalazione
Presentazione di un eMDR. Inizialmente, per effettuare una presentazione, è necessario disporre di una firma elettronica e assicurarsi che i nomi dei file di presentazione includano un solo punto, utilizzato per indicare l'estensione del tipo di file (ad esempio 555.xml o 555.pdf). Tuttavia, i tempi di consegna ed elaborazione della domanda dipendono dalla dimensione complessiva della presentazione; le presentazioni più grandi richiedono più tempo per essere consegnate ed elaborate.
Fase 2: Gateway per le Presentazioni Elettroniche (ESG)
Quando la tua presentazione raggiunge l'ESG, dovresti ricevere rapidamente una conferma #1 a meno che l'ESG non sia inattivo per manutenzione. Sei tenuto a controllare lo stato del tuo MDR sul sito web dell'ESG.
Fase 3: CRDH
L'eMDR viene instradato automaticamente dall'ESG al Centro per i Dispositivi e la Salute Radiologica (CDRH). Una volta instradato, come nel passaggio 2, si dovrebbe ricevere una conferma di ricezione, ovvero il #2.
Fase 4: Esperienza del Dispositivo da Parte del Fabbricante e dell'Utilizzatore (MAUDE)
Quando il CDRH convalida e aggiorna la presentazione nel database degli eventi avversi (MAUDE), si prevede che il mittente riceva una conferma #3. Si noti che eventuali errori che si verificano durante la convalida e il caricamento vengono registrati.
La Segnalazione di Dispositivi Medici (MDR) è un processo fondamentale che contribuisce a salvare vite e a proteggere i pazienti da rischi inutili. Garantisce che tutte le parti coinvolte nell'assistenza ai pazienti siano responsabili e attente nell'uso dei dispositivi.
L'eMDR semplifica la segnalazione, ma la documentazione e il follow-up possono richiedere molte risorse. Fatelo bene la prima volta; contattateci all'indirizzo sales@freyrsolutions.com.