2 minuty czytania
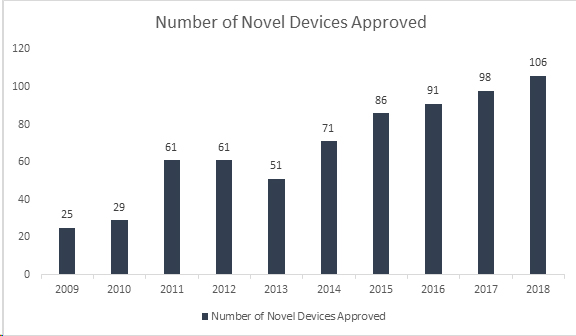
Czy wiesz, że? Amerykańska Agencja ds. Żywności i Leków (FDA) ustanowiła kolejny rekord, zatwierdzając 106 nowych wyrobów w 2018 roku, czyniąc go najbardziej udanym rokiem dla wyrobów medycznych. Dzięki temu osiągnięciu FDA przekroczyła swój 40-letni rekord ustanowiony w 2017 roku, zatwierdzając 99 nowych wyrobów, wykazując ciągły wzrost przez ostatnie 8 lat. Zatwierdzone wyroby obejmują szereg innowacyjnych produktów, takich jak automatyczne systemy dawkowania insuliny dla dzieci, najmniejsza na świecie zastawka serca dla noworodków, pierwsza mobilna aplikacja medyczna pomagająca w zarządzaniu zaburzeniami związanymi z używaniem opioidów i substancji psychoaktywnych oraz technologie sztucznej inteligencji, które diagnozują retinopatię cukrzycową.
FDA zawsze promowała bezpieczeństwo i innowacyjność wyrobów medycznych, aby zapewnić ich wysoką jakość. Aby sprostać rosnącej liczbie zatwierdzeń nowych wyrobów i zapewnić ich bezpieczeństwo, FDA planuje „zmodernizować” ścieżkę dopuszczania wyrobów medycznych. Według agencji, modernizacja procesu dopuszczania może wymagać nowych uprawnień. W 2018 roku FDA i Centrum ds. Wyrobów i Zdrowia Radiologicznego (CDRH) opublikowały wspólny dokument, w którym stwierdzono, że 510(k) był jednym z dwóch rodzajów zgłoszeń dodanych do definicji nowych wyrobów. Ponadto, zwolnienia dla wyrobów humanitarnych (HDE) również zostały dodane do definicji nowego wyrobu po zmianach wprowadzonych w programie CDRH dotyczącym przełomowych wyrobów medycznych, wynikających z Ustawy o XXI wieku (21st Century Act).
Propozycja FDA i CDRH jest odpowiedzią na potencjalną potrzebę dążenia do nowych uprawnień w celu modernizacji procesu 510(k). Celem propozycji jest ograniczenie stosowania urządzeń referencyjnych, uznawanych za Zasadniczo Równoważne (SE), które są starsze niż 10 lat, w celu promowania innowacji. Jest to krok naprzód w odniesieniu do wytycznych opublikowanych przez agencję w kwietniu 2018 roku. Projekt został opublikowany przez FDA w celu zaproponowania rozszerzenia skróconego programu 510(k) w CDRH FDA pod tytułem – „Ścieżka oparta na bezpieczeństwie i wydajności”. Został wprowadzony w celu zmniejszenia obciążeń związanych z przepisami dotyczącymi wyrobów medycznych. Podejście to ma również na celu zwiększenie efektywności przeglądu zgłoszeń 510(k), co zmniejsza presję na agencję.
Oto niektóre z najważniejszych punktów wytycznych:
- Nowa ścieżka ocenia bezpieczeństwo i skuteczność urządzeń w oparciu o ustalone standardy bezpieczeństwa i wskaźniki wydajności.
- Pomimo nowych standardów, wyroby będą musiały być zgodne z istniejącymi standardami, aby mogły być wprowadzane do obrotu.
- Nowoczesna technologia zostanie przetestowana zgodnie z nowoczesnymi standardami
- Podejście to przyczyni się do większej konkurencji w rozwoju bezpieczniejszych urządzeń.
Liczba wyrobów medycznych zgłaszanych do zatwierdzenia wzrosła wykładniczo na przestrzeni lat. Dało to FDA szerokie pole do przyjęcia i wdrożenia innowacyjnych środków w celu usprawnienia ścieżek zatwierdzania. Agencja mocno wierzy w zalety tej propozycji, ale odpowiedź branży wciąż czeka na rozszyfrowanie.
W związku z tym, że FDA nieustannie opracowuje nowe dokumenty wytyczne w celu uaktualnienia rejestracji wyrobów medycznych, konieczne jest, aby producenci wyrobów medycznych śledzili je i odpowiednio działali. Bądź na bieżąco. Zachowaj zgodność.









