
3 minuty czytania
Zastosowanie technologii oprogramowania w różnorodnym zarządzaniu opieką zdrowotną, w tym w diagnozowaniu lub leczeniu stanów chorobowych, przyspiesza w niespotykanym tempie. Globalne organy ds. wyrobów medycznych zmieniają swoje przepisy i wytyczne, aby sprostać tym dynamicznie rozwijającym się technologiom wyrobów. W Australii, Therapeutic Goods Administration (TGA) jest organem regulacyjnym odpowiedzialnym za nadzór nad wyrobami medycznymi, w tym oprogramowaniem i aplikacjami mobilnymi. W styczniu 2021 roku TGA opublikowała pierwotny projekt przepisów dotyczących oprogramowania wyrobów medycznych, który został zmieniony w lutym 2021 roku. 27 lipca 2021 roku TGA opublikowała szczegółowy schemat blokowy, który rozwiewa typowe niejasności, jakie producenci wyrobów i specjaliści ds. Spraw regulacyjnych mogą mieć w związku z klasyfikacją oprogramowania wyrobów medycznych.
TGA identyfikuje oprogramowanie używane w dziedzinie wyrobów medycznych jako Oprogramowanie jako wyrób medyczny (SaMD), Oprogramowanie wbudowane w wyrób medyczny (SiMD) oraz Oprogramowanie sterujące wyrobami medycznymi. Według TGA, termin Oprogramowanie jako wyrób medyczny (lub SaMD) odnosi się do każdego oprogramowania, które może działać na laptopie, smartfonie lub tablecie, a którego przeznaczenie mieści się w normach wyrobu medycznego. Natomiast SiMD to oprogramowanie, które jest integralną częścią wyrobu i jest zazwyczaj ważne dla jego prawidłowego funkcjonowania. Niektóre oprogramowania sterują wyrobami medycznymi fizycznie lub za pośrednictwem połączeń bezprzewodowych, takich jak Bluetooth, Wi-Fi itp. TGA reguluje wszystkie trzy (03) typy oprogramowania.
Niewiele oprogramowania wyrobów medycznych jest wyłączonych lub zwolnionych z regulacji TGA dotyczącej oprogramowania jako wyrobu medycznego w oparciu o zasady dostosowania regulacji do przepisów międzynarodowych i zmniejszenia obciążeń poprzez nieregulowanie produktów, w przypadku braku poważnego ryzyka dla bezpieczeństwa oraz gdy istnieją już odpowiednie struktury lub ramy kontroli produktu lub systemu.
| Zwolnione urządzenia programowe | Wyłączone urządzenia programowe |
|---|---|
Np., Niewiele systemów wspomagania decyzji klinicznych, które spełniają trzy główne kryteria: 1. Jeśli oprogramowanie nie jest przeznaczone do analizowania ani przetwarzania obrazu medycznego. 2. Oprogramowanie ma na celu jedynie zapewnienie wsparcia/rekomendacji pracownikowi służby zdrowia. 3. Oprogramowanie nie ma na celu zastępowania oceny klinicznej specjalisty. | Produkty, które nie wchodzą w kategorię wyrobów medycznych i nie podlegają żadnym wymogom regulacyjnym TGA, są nazywane wyłączonymi wyrobami oprogramowania. Np.,
|
TGA zrewidowała regulacje dotyczące wyrobów medycznych zawierających oprogramowanie, które steruje innymi wyrobami medycznymi lub z nimi współdziała, zarówno zewnętrznie, jak i wewnętrznie, oraz oprogramowania, które samo w sobie funkcjonuje jako wyrób medyczny. Główne zmiany obejmują wprowadzenie nowych zasad klasyfikacji, wyjaśnienie granic regulowanych produktów oprogramowania, a także aktualizację podstawowych zasad w celu sprecyzowania wymagań dla wyrobów medycznych opartych na oprogramowaniu.
Wraz z wprowadzeniem nowych zasad klasyfikacji, niektóre wyroby medyczne oparte na oprogramowaniu są przeklasyfikowywane do wyższych klas ryzyka i muszą przejść odpowiednie procedury oceny zgodności. Sponsorzy i producenci takich przeklasyfikowanych wyrobów muszą być wpisani do ARTG (Australijskiego Rejestru Produktów Leczniczych) przed 25 lutego 2021 r. Producenci i sponsorzy, którzy już złożyli wnioski do TGA przed 25 lutego 2021 r., muszą złożyć formularz zgłoszeniowy do TGA.
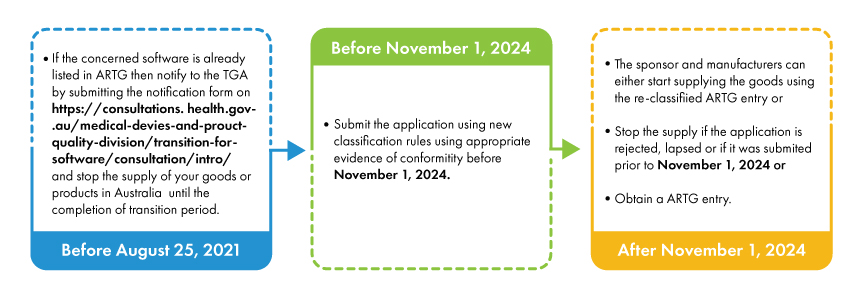
Klasyfikacja oprogramowania wyrobów medycznych w Australii:
Producenci muszą określić zamierzone zastosowanie SaMD, co pomoże w dalszych procedurach klasyfikacji i oceny zgodności. Nowe rozporządzenie wprowadza również nowe zasady klasyfikacji dla programowanych i programowalnych wyrobów medycznych lub wyrobów medycznych będących oprogramowaniem, a klasa oprogramowania będzie zależeć od ciężkości monitorowanej/leczonej choroby.
![]()
Zmiany w zasadniczych wymaganiach:
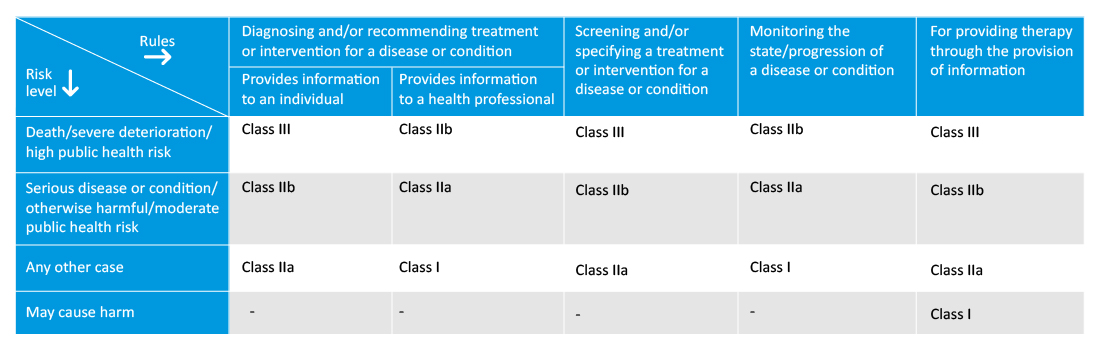
Poniżej przedstawiono zmiany wprowadzone do zasadniczych wymagań, obowiązujące od 25 lutego 2021, dla wyrobu medycznego opartego na oprogramowaniu.
Zmiany wprowadzone w przepisach dotyczących oprogramowania przez TGA mogą zmniejszyć obciążenie producentów związane z kategoryzacją ich oprogramowania jako regulowanego lub nieregulowanego i mogą pomóc w terminowym przygotowaniu dokumentów wymaganych do oceny zgodności TGA.
Po rozszyfrowaniu wszystkich scenariuszy, czy dążysz do umieszczenia swojego oprogramowania w Australijskim Rejestrze Produktów Leczniczych (ARTG) z mniejszym obciążeniem? W celu umieszczenia w ARTG wyrobów medycznych, IVD lub SaMD, skontaktuj się z ekspertem. Bądź na bieżąco. Bądź zgodny z przepisami.









