

3 minuty czytania
W ewoluującym świecie wyrobów medycznych, zgodność nie jest jednorazowym zadaniem — to ciągłe zobowiązanie. Ciągłe monitorowanie i aktualizacje kluczowych raportów, takich jak Raporty z Oceny Klinicznej (CER), Raporty z Oceny Działania (PER) oraz Okresowe Raporty z Bezpieczeństwa (PSUR), są kluczowe przez cały cykl życia wyrobu medycznego, od początkowych badań do nadzoru po wprowadzeniu do obrotu. Ponieważ krajobraz postępów medycznych i wymagań regulacyjnych stale się zmienia, zapewnienie bezpieczeństwa i zgodności wyrobów medycznych i IVD poprzez skuteczne pisanie medyczne pozostaje kamieniem węgielnym sukcesu i długoterminowej rentowności.
Przyjrzyjmy się, jak zarządzanie cyklem życia pozostaje kluczowe dla sukcesu wyrobu medycznego.
Wprowadzenie wyrobu medycznego na rynek to ukoronowanie lat wysiłków poświęconych na kilka etapów, takich jak badania, rozwój, badania kliniczne, zgłoszenia regulacyjne i nadzór po wprowadzeniu do obrotu. Proces ten trwa wiele lat, a każdy etap gromadzi duże ilości kluczowych danych, które muszą być starannie zebrane i przeanalizowane, aby zapewnić bezpieczeństwo wyrobu.
Zgodność nie jest jednorazowym działaniem, a skuteczne zarządzanie cyklem życia pomaga uniknąć opóźnień, które mogą być kosztowne w przyszłości. Opóźnienia te często wynikają z nieskutecznego planowania nieprzewidzianych zmian regulacyjnych lub braku zgodności, które mogły zostać przeoczone. Regularne aktualizacje kluczowych dokumentów, takich jak CER, PER i PSUR, zapewniają, że producenci spełniają zmieniające się wymogi regulacyjne i utrzymują bezpieczeństwo produktu. Zarządzanie cyklem życia zapewnia, że producenci od samego początku zaplanowali każdy etap, dzięki czemu mogą uniknąć wszelkich pułapek i zapewnić udane wejście na rynek oraz długowieczność swoich wyrobów.
Czy jesteś na bieżąco z wymogami zgodności?
Od bandaży po implanty, branża wyrobów medycznych to nowoczesny, kreatywny sektor z bogactwem perspektyw. Pomimo silnego pragnienia producentów, aby dostarczać na rynek bezpieczne i wysokiej jakości produkty, nadal istnieje niejasność. EU MDR zastąpiło Dyrektywę o wyrobach medycznych (MDD) i Dyrektywę o aktywnych wyrobach medycznych do implantacji (AIMDD). Rozporządzenie to zostało wdrożone w celu narzucenia bardziej rygorystycznych kontroli, poprawy bezpieczeństwa pacjentów i promowania większej przejrzystości w sektorze opieki zdrowotnej. Istnieje wiele niejasności dotyczących tych standardów, a kluczowe kwestie zgodności są często pomijane. Jednak producenci wyrobów medycznych powinni podjąć kroki, aby uniknąć trudności regulacyjnych.
Zbadajmy najczęstsze wyzwania stojące przed producentami wyrobów medycznych.
- Utrzymywanie CAPA (Corrective and Preventive Action)
- Przestrzeganie Procedur Rozpatrywania Skarg
- Procedury nadzoru
- Ocena Kliniczna i Nadzór po wprowadzeniu do obrotu
- Współpraca z jednostkami notyfikowanymi
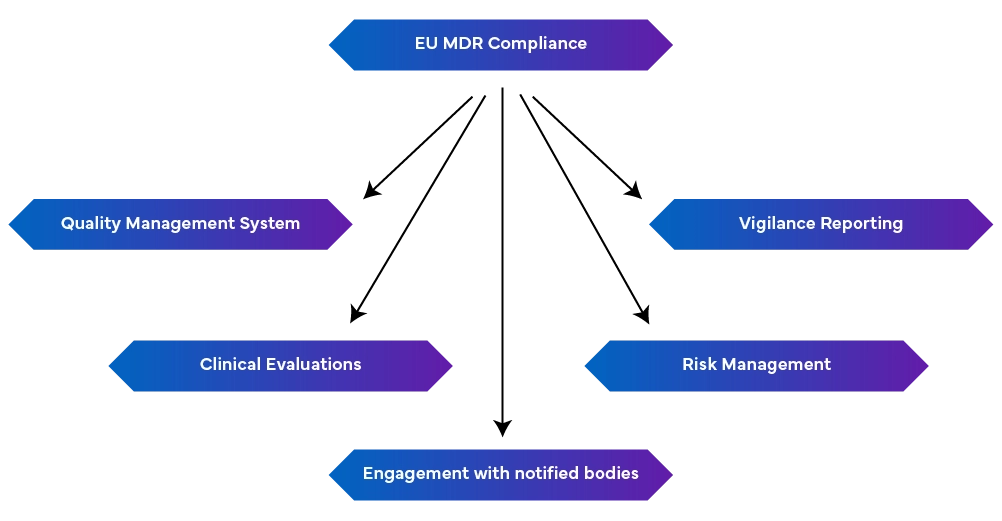
Najlepsze praktyki w celu zachowania zgodności
Aby sprostać tym wyzwaniom, w Freyr stworzyliśmy
-solidny System Zarządzania Jakością (EN ISO 13485:2016): Obejmuje to aspekty produkcyjne, w tym zgodność regulacyjną, dokumentację techniczną, deklaracje zgodności UE oraz zarządzanie ryzykiem.
-Raportowanie Czujności Farmakoterapeutycznej: Utrzymywanie raportowania czujności farmakoterapeutycznej jako ciągłego procesu, a nie jednorazowego działania.
- Proaktywne Zarządzanie Ryzykiem: Polega na identyfikowaniu i łagodzeniu ryzyka w całym cyklu życia wyrobu poprzez regularne aktualizacje w celu reagowania na pojawiające się zagrożenia.
- Przeprowadzanie Ocen Klinicznych i Nadzoru Po Wprowadzeniu do Obrotu: Odbywa się to poprzez wykazanie bezpieczeństwa i działania wyrobu na podstawie danych klinicznych. W razie potrzeby należy przeprowadzić badania kliniczne, a oceny kliniczne powinny być regularnie aktualizowane danymi z nadzoru po wprowadzeniu do obrotu. Wymagane są również okresowe raporty z aktualizacji bezpieczeństwa podsumowujące ustalenia.
- Kontakt z Jednostkami Notyfikowanymi: Zmiany w przepisach mogą wpływać na wymagania MDR, dlatego kluczowe jest utrzymywanie dobrych relacji z Jednostkami Notyfikowanymi w celu uzyskiwania szybkich aktualizacji i modyfikacji.
Jak ekspert ds. regulacji może Ci pomóc
Nadążanie za ciągle zmieniającym się krajobrazem regulacyjnym może być trudne. Dlaczego więc nie pozwolić ekspertowi poprowadzić Państwa przez ten labirynt? Zarządzanie wymogami regulacyjnymi w całym cyklu życia wyrobu medycznego jest nie tylko złożone, ale także wymaga dużych zasobów. W przypadku mniejszych firm, gdzie wewnętrzne zasoby mogą być lepiej wykorzystane w innych obszarach, pomoc zewnętrznego partnera regulacyjnego może okazać się nieoceniona. Oferują oni specjalistyczną wiedzę na temat ewoluujących przepisów i zapewniają, że Państwa kluczowe raporty – takie jak CER, PER i PSUR – są stale aktualizowane i zgodne z wymogami.
Możesz uniknąć kosztownych opóźnień, zapobiec niezgodnościom i usprawnić proces zatwierdzania. Dodatkowo, ekspert regulacyjny może zaoferować rozwiązania dostosowane do specyficznych potrzeb Twojego urządzenia, zapewniając aktualność nadzoru po wprowadzeniu na rynek, analizy luk i całej dokumentacji zgodności. Ich zaangażowanie może uprościć cały proces, zminimalizować ryzyko i zapewnić, że urządzenie spełni oczekiwania dotyczące bezpieczeństwa i wydajności.
Freyr może pomóc Państwu we wszystkich potrzebach regulacyjnych dotyczących zarządzania cyklem życia produktu. Skontaktuj się z nami już dziś!









