

3 min de leitura
Realizar um ensaio clínico é um passo importante no desenvolvimento de um medicamento ou dispositivo médico. É necessária uma submissão de Pedido de Autorização de Ensaio Clínico (CTA) para que as empresas farmacêuticas ou promotores obtenham a aprovação das autoridades regulamentares para iniciar estudos em humanos. À medida que a investigação clínica se torna mais global e complexa, compreender as práticas internacionais em evolução, os passos críticos da submissão e as vantagens oferecidas por um(s) parceiro(s) de Assuntos Regulamentares é crucial tanto para promotores como para organizações de investigação.
Pedido de Autorização de Ensaio Clínico (CTA) e as suas Secções:
Um Pedido de Autorização de Ensaio Clínico (CTA) é o dossiê regulamentar que os promotores devem submeter às autoridades regulamentares nacionais (NRAs) antes de iniciar um estudo clínico envolvendo participantes humanos. Os CTAs são obrigatórios para a maioria dos ensaios intervencionais de medicamentos, dispositivos ou produtos biológicos. Garantem que o ensaio é cientificamente sólido, eticamente apropriado e alinhado com as diretrizes locais e internacionais — tais como as normas de Boas Práticas Clínicas (GCP) do Conselho Internacional para a Harmonização (ICH) e os requisitos nacionais.
As secções principais num CTA típico incluem (mas não se limitam a):
- Carta de apresentação e formulários de submissão
- Protocolo do estudo clínico e brochura do investigador
- Dossiê do produto e evidência de Boas Práticas de Fabrico (GMP)
- Aprovações de ética e do Comité de Ética em Investigação (IRB)
- Documentação de seguro e proteção dos participantes
- Plano de monitorização da segurança dos dados e declarações financeiras
Práticas Globais em Submissões de CTA
Os processos e requisitos de CTA diferem entre regiões, mas vários princípios comuns estão a emergir:
1. Harmonização de Formatos e Normas
- Muitos países utilizam o Documento Técnico Comum (CTD) do ICH, simplificando as submissões de estudos multinacionais. A sua estrutura modular apoia a revisão paralela e simplifica as atualizações.
- As adaptações locais são comuns, como nas regiões da Ásia-Pacífico e Pacífico Ocidental, onde os países frequentemente combinam o CTD com formulários nacionais, feedback de revisão, necessidades linguísticas ou estudos de ponte.
2. Consulta Regulamentar
- O envolvimento precoce com as agências — como a FDA dos US, as autoridades nacionais da UE e as agências no Japão, China e mercados emergentes chave — é encorajado, especialmente para ensaios clínicos globais e multirregionais (MRCTs). As reuniões de aconselhamento científico reduzem futuras rejeições e orientam a otimização do protocolo.
3. Vias Colaborativas e Aceleradas
- As agências aceitam cada vez mais a “confiança” ou avaliações colaborativas, onde aproveitam revisões ou aprovações por autoridades regulamentares rigorosas, aumentando a velocidade e a consistência.
- Opções de revisão acelerada ou prioritária podem estar disponíveis, especialmente durante emergências de saúde ou para terapias inovadoras.
4. Integração da Revisão Ética
- Na UE e em muitas outras regiões, as revisões éticas e regulamentares podem ocorrer em paralelo ou através de plataformas coordenadas para evitar submissões separadas e encurtar os tempos de arranque.
5. Transparência e Publicação
- É agora padrão registar ensaios em bases de dados reconhecidas antes da aprovação, e muitos países reportam publicamente os estados de aprovação de CTA, contribuindo para a transparência global e as melhores práticas.
Como Pode um Parceiro Como a Freyr Ajudar?
Um parceiro de Assuntos Regulamentares (RA) de confiança como a Freyr oferece apoio contínuo e mitigação de riscos ao longo de todo o processo de CTA. Eis porque o envolvimento de um especialista como este prepara os patrocinadores para o sucesso:
1. Global Regulatory intelligence Atualizada
- Monitorizamos continuamente as alterações regulamentares, as interpretações regionais e as práticas de submissão em todo o mundo. Isto garante que cada CTA é elaborada com base nos requisitos mais atuais, minimizando questões, rejeições e atrasos dispendiosos nos ensaios.
2. Preparação e Revisão de Dossiês
- Os especialistas da Freyr desenvolvem e compilam CTAs utilizando os mais recentes formatos ICH CTD e modelos nacionais adaptados. Coordenam as traduções de documentos, organizam as respostas do dossiê e garantem qualidade uniforme e conformidade com as GCP para cada submissão.
3. Consulta Regulamentar e Ligação com Agências
- A Freyr pode representar os patrocinadores em reuniões de aconselhamento científico pré-CTA, gerir questões das agências após a submissão e apoiar as comunicações com as autoridades e os Comités de Ética.
4. Gestão Global de Projetos
- Com equipas multiculturais experientes e ferramentas robustas de gestão de projetos, a Freyr coordena submissões simultâneas, acompanha o progresso país a país e promove o alinhamento para MRCTs (Ensaios Clínicos Multirregionais).
5. Estratégia de Dependência e Revisão Acelerada
- A Freyr ajuda os promotores a alavancar estruturas de confiança, a preparar dossiês abreviados para revisões dispensadas ou aceleradas, e a alinhar os dados de submissão para máxima aceitação regulamentar.
6. Preparação para Auditorias e Controlo de Alterações
- O parceiro garante que a documentação e os procedimentos dos promotores cumprem os padrões de auditoria, facilitando uma aprovação regulamentar rápida e inspeções sem problemas.
Conclusão
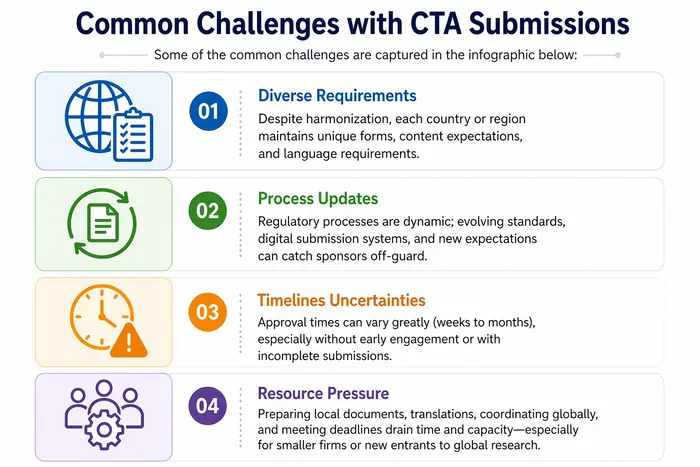
A submissão de CTA é uma porta de entrada complexa, mas essencial, para a investigação clínica, com requisitos globais e específicos da região. As tendências globais incluem a harmonização do CTD, a revisão paralela de ética e regulamentar, mecanismos de confiança e novas normas de transparência. Os promotores enfrentam desafios notáveis no cumprimento de requisitos diversos, no acompanhamento das mudanças regulamentares e na gestão de prazos globais. Neste cenário, um parceiro de Assuntos Regulamentares como a Freyr oferece experiência End-to-End em inteligência regulamentar, preparação de dossiês, interações com agências, vias aceleradas e preparação para auditorias — aumentando substancialmente as probabilidades de inícios de ensaios clínicos atempados e bem-sucedidos.









